Frontiers in Genetics (Čeština)
Úvod
Promotory jsou klíčové prvky, které patří do nekódujících oblastí v genomu. Do značné míry řídí aktivaci nebo represi genů. Jsou umístěny v blízkosti a proti směru transkripčního počátečního místa genu (TSS). Oblast lemující promotor genu může obsahovat mnoho klíčových krátkých DNA prvků a motivů (5 a 15 bází dlouhých), které slouží jako rozpoznávací místa pro proteiny, které poskytují správná iniciace a regulace transkripce downstream genu (Juven-Gershon et al., 2008). Zahájení transkriptu genu je nejzákladnějším krokem v regulaci genové exprese. Jádro promotoru je minimální úsek DNA sekvence, který tvoří TSS a dostatečný k přímé iniciaci transkripce. Délka promotoru jádra se obvykle pohybuje mezi 60 a 120 páry bází (bp).
TATA-box je subsekvence promotoru, která označuje další molekuly, kde transkripce začíná. Byl pojmenován „TATA-box“, protože jeho sekvence je charakterizována opakováním párů bází T a A (TATAAA) (Baker et al., 2003). Drtivá většina studií na TATA-boxu byla provedena na lidech, kvasinkách, a Drosophila genom, nicméně podobné prvky byly nalezeny u jiných druhů, jako jsou archaea a starověké eukaryoty (Smale a Kadonaga, 2003). V případě člověka má 24% genů promotorové oblasti obsahující TATA-box (Yang et al., 2007 ). U eukaryot se TATA-box nachází přibližně 25 bp před TSS (Xu et al., 2016). Je schopen definovat směr transkripce a také označuje řetězec DNA, který se má číst. Proteiny nazývané transkripční faktory váže se na několik nekódujících oblastí, včetně TATA-boxu, a získává enzym zvaný RNA polymeráza, který syntetizuje RNA z DNA.
Vzhledem k důležité roli promotorů v transkripci genů se stává přesnou predikcí promotorových míst požadovaný krok v genové expresi, interpretaci vzorů a vytváření a porozumění funkčnost genetických regulačních sítí. Existovaly různé biologické experimenty pro identifikaci promotorů, jako je mutační analýza (Matsumine et al., 1998) a imunoprecipitační testy (Kim et al., 2004; Dahl a Collas, 2008). Tyto metody však byly nákladné i časově náročné. V poslední době, s vývojem sekvenování nové generace (NGS) (Behjati a Tarpey, 2013), bylo sekvenováno více genů různých organismů a jejich genové prvky byly výpočtově prozkoumány (Zhang et al., 2011). Na druhou stranu inovace technologie NGS vedla k dramatickému poklesu nákladů na sekvenování celého genomu, takže je k dispozici více dat o sekvenování. Dostupnost dat láká výzkumné pracovníky k vývoji výpočetních modelů pro predikční úlohu promotoru. Stále však jde o neúplný úkol a neexistuje žádný efektivní software, který by mohl přesně předvídat promotéry.
Prediktory promotorů lze kategorizovat na základě využívaného přístupu do tří skupin, a to na základě signálu, přístupu založeného na obsahu a přístup založený na GpG. Prediktory založené na signálu se zaměřují na promotorové prvky související s vazebným místem RNA polymerázy a ignorují neelementové části sekvence. Výsledkem bylo, že přesnost predikce byla slabá a neuspokojivá. Mezi příklady prediktorů založených na signálu patří: PromoterScan (Prestridge, 1995), který používal extrahované vlastnosti TATA-boxu a vážené matice vazebných míst transkripčního faktoru s lineárním diskriminátorem ke klasifikaci promotorových sekvencí z nepromotorových; Promoter2.0 (Knudsen, 1999), který extrahoval vlastnosti z různých boxů jako TATA-Box, CAAT-Box a GC-Box a předal je umělým neuronovým sítím (ANN) ke klasifikaci; NNPP2.1 (Reese, 2001), který využil iniciátorový prvek (Inr) a TATA-Box pro extrakci funkcí a neuronovou síť s časovým zpožděním pro klasifikaci, a Down a Hubbard (2002), kteří použili TATA-Box a využili relevantní vektorové stroje (RVM) jako klasifikátor. Prediktory založené na obsahu se spoléhaly na počítání frekvence k-mer spuštěním okna k-délky přes sekvenci. Tyto metody však ignorují prostorové informace párů bází v sekvencích. Mezi příklady prediktorů založených na obsahu patří: PromFind (Hutchinson, 1996), který používal k-mer frekvenci k provedení predikce hexamerového promotoru; PromoterInspector (Scherf et al., 2000), který identifikoval oblasti obsahující promotory na základě společného genomového kontextu promotorů polymerázy II skenováním specifických rysů definovaných jako motivy s proměnnou délkou; MCPromoter1.1 (Ohler et al., 1999), který k predikci promotorových sekvencí používal jediný interpolovaný Markovův řetězec (IMC) 5. řádu.Nakonec prediktory založené na GpG využily umístění ostrovů GpG jako promotorové oblasti nebo první oblasti exonu v lidských genech obvykle obsahují ostrovy GpG (Ioshikhes a Zhang, 2000; Davuluri et al., 2001; Lander et al., 2001; Ponger a Mouchiroud, 2002). Pouze 60% promotorů však obsahuje ostrovy GpG, proto přesnost predikce tohoto druhu prediktorů nikdy nepřesáhla 60%.
V poslední době byly pro predikci promotorů použity přístupy založené na sekvenci. Yang a kol. (2017) využili různé strategie extrakce funkcí k zachycení nejdůležitějších informací o sekvenci za účelem předpovědi interakcí zesilovače s promotorem. Lin a kol. (2017) navrhli pro identifikaci promotoru sigma70 v prokaryotě sekvenční prediktor s názvem „iPro70-PseZNC“. Podobně Bharanikumar et al. (2018) navrhli PromoterPredict za účelem předpovědi síly promotorů Escherichia coli na základě přístupu dynamické vícenásobné regrese, kde byly sekvence reprezentovány jako matice polohové hmotnosti (PWM). Kanhere a Bansal (2005) využili rozdíly ve stabilitě sekvence DNA mezi promotorovými a nepropagačními sekvencemi k jejich rozlišení. Xiao a kol. (2018) představili dvouvrstvý prediktor zvaný iPSW (2L) -PseKNC pro identifikaci promotorových sekvencí i sílu promotorů extrakcí hybridních rysů ze sekvencí.
Všechny výše uvedené prediktory vyžadují doménu znalosti, aby bylo možné tyto funkce ručně zpracovat. Na druhé straně přístupy založené na hlubokém učení umožňují vytváření efektivnějších modelů pomocí přímých dat (sekvence DNA / RNA) přímo. Hluboká konvoluční neuronová síť dosáhla nejmodernějších výsledků v náročných úkolech, jako je zpracování obrazu, videa, zvuku a řeči (Krizhevsky et al., 2012; LeCun et al., 2015; Schmidhuber, 2015; Szegedy et al. , 2015). Kromě toho byl úspěšně aplikován v biologických problémech, jako je DeepBind (Alipanahi et al., 2015), DeepCpG (Angermueller et al., 2017), výběr pobočkových bodů (Nazari et al., 2018), predikce alternativních spojovacích míst (Oubounyt et al., 2018), predikce 2 „-methylačních míst (Tahir et al., 2018), kvantifikace sekvence DNA (Quang a Xie, 2016), subcelulární lokalizace lidského proteinu (Wei et al., 2018) atd. Dále CNN nedávno získala významnou pozornost v úloze rozpoznávání promotorů. Velmi nedávno Umarov a Solovyev (2017) představili CNNprom pro rozlišení krátkých promotorových sekvencí, tato architektura založená na CNN dosáhla vysokých výsledků při klasifikaci promotorových a nepropagačních sekvencí. Poté byl tento model vylepšen autor Qian et al. (2018), kde autoři použili klasifikátor podpory vektorových strojů (SVM) ke kontrole nejdůležitějších prvků promotorové sekvence. Nejvlivnější prvky byly ponechány nekomprimované při komprimaci těch méně důležitých. Tento proces vedl k lepšímu výkonu. Nedávno navrhli model identifikace dlouhého promotoru Umarov et al. (2019), ve kterém se autoři zaměřili na identifikaci polohy TSS.
Ve všech výše zmíněných pracích byla negativní sada extrahována z nepropagačních oblastí genomu. S vědomím, že promotorové sekvence jsou bohaté výhradně na specifické funkční prvky, jako je TATA-box, který je umístěn na –30 ~ –25 bp, GC-Box, který je umístěn na –110 ~ –80 bp, CAAT-Box, který je na – 80 ~ –70 bp atd. To má za následek vysokou přesnost klasifikace kvůli velkému rozdílu mezi pozitivními a negativními vzorky z hlediska struktury sekvence. Úkol klasifikace je navíc snazší dosáhnout, například modely CNN se při rozhodování o typu sekvence budou spoléhat pouze na přítomnost nebo nepřítomnost některých motivů v jejich konkrétních pozicích. Tyto modely tedy mají velmi nízkou přesnost / citlivost (vysoce falešně pozitivní), když jsou testovány na genomových sekvencích, které mají promotorové motivy, ale nejedná se o promotorové sekvence. Je dobře známo, že v genomu je více motivů TATAAA než těch, které patří do promotorových oblastí. Například samotná sekvence DNA lidského chromozomu 1, ftp://ftp.ensembl.org/pub/release-57/fasta/homo_sapiens/dna/, obsahuje 151 656 motivů TATAAA. Je to více než přibližný maximální počet genů v celém lidském genomu. Pro ilustraci tohoto problému si všimneme, že při testování těchto modelů na nepropagačních sekvencích, které mají TATA-box, špatně klasifikují většinu těchto sekvencí. Proto, aby bylo možné vygenerovat robustní klasifikátor, měla by být záporná sada vybrána opatrně, protože určuje funkce, které bude používat klasifikátor za účelem diskriminace tříd. Důležitost této myšlenky byla prokázána v předchozích pracích, jako je (Wei et al., 2014). V této práci se věnujeme hlavně této problematice a navrhujeme přístup, který integruje některé funkční motivy pozitivní třídy do negativní třídy, aby se snížila závislost modelu na těchto motivech.Využíváme CNN v kombinaci s modelem LSTM k analýze sekvenčních charakteristik lidských a myších TATA a jiných než TATA eukaryotických promotorů a vytváření výpočetních modelů, které dokážou přesně rozlišit krátké promotorové sekvence od nepropagačních.
Materiály a metody
2.1. Datová sada
Datové sady, které se používají k trénování a testování navrhovaného prediktoru promotoru, se shromažďují od lidí a myší. Obsahují dvě charakteristické třídy promotorů, jmenovitě promotory TATA (tj. Sekvence, které obsahují TATA-box) a promotory jiné než TATA. Tyto datové sady byly vytvořeny z databáze Eukaryotic Promoter Database (EPDnew) (Dreos et al., 2012). EPDnew je nová sekce pod dobře známým souborem dat EPD (Périer et al., 2000), kde je anotována neredundantní sbírka eukaryotických promotorů POL II, kde bylo počáteční místo transkripce stanoveno experimentálně. Poskytuje vysoce kvalitní promotory ve srovnání s kolekcí promotorů ENSEMBL (Dreos et al., 2012) a je veřejně přístupný na https://epd.epfl.ch//index.php. Z EPDnew jsme pro každý organismus stáhli genomové sekvence promotoru TATA a non-TATA. Tato operace vyústila v získání čtyř datových sad promotorů, konkrétně: Human-TATA, Human-non-TATA, Mouse-TATA a Mouse-non-TATA. Pro každý z těchto datových souborů je vytvořena negativní sada (nepropagační sekvence) se stejnou velikostí pozitivní na základě navrhovaného přístupu, jak je popsáno v následující části. Podrobnosti o počtech promotorových sekvencí pro každý organismus jsou uvedeny v tabulce 1. Všechny sekvence mají délku 300 bp a byly extrahovány z -249 ~ + 50 bp (+1 označuje TSS pozici). Jako kontrolu kvality jsme k posouzení navrhovaného modelu použili pětinásobnou křížovou validaci. V tomto případě se pro trénink používá 3krát, pro ověření se použije 1krát a pro testování se použije zbytek. Navrhovaný model je tedy trénován 5krát a je vypočítán celkový výkon pětinásobku.

Tabulka 1. Statistiky čtyř souborů dat použitých v této studii.
2.2. Negativní konstrukce datové sady
Abychom mohli trénovat model, který dokáže přesně provést klasifikaci promotorových a nepromotorových sekvencí, musíme opatrně zvolit negativní sadu (nepropagační sekvence). Tento bod je zásadní při vytváření modelu schopného dobře zobecnit, a proto je schopen udržovat jeho přesnost při hodnocení na náročnějších souborech dat. Předchozí práce, jako například (Qian et al., 2018), konstruovaly negativní množinu náhodným výběrem fragmentů z genomových nepropagačních oblastí. Je zřejmé, že tento přístup není zcela rozumný, protože pokud neexistuje průnik mezi kladnými a zápornými množinami. Model tedy snadno najde základní funkce pro oddělení obou tříd. Například motiv TATA lze nalézt ve všech pozitivních sekvencích na konkrétní pozici (obvykle 28 bp před TSS, v našem souboru dat mezi –30 a –25 pb). Náhodné vytváření záporné množiny, která tento motiv neobsahuje, proto v této datové sadě vyprodukuje vysoký výkon. Model však selhává při klasifikaci negativních sekvencí, které mají TATA motiv jako promotory. Stručně řečeno, hlavní chyba v tomto přístupu spočívá v tom, že při trénování modelu hlubokého učení se pouze naučí rozlišovat pozitivní a negativní třídy na základě přítomnosti nebo nepřítomnosti některých jednoduchých funkcí na konkrétních pozicích, což znemožňuje tyto modely. V této práci se snažíme vyřešit tento problém vytvořením alternativní metody pro odvození záporné množiny od kladné.
Naše metoda je založena na skutečnosti, že kdykoli jsou vlastnosti společné mezi záporným a pozitivní třída, model má při rozhodování tendenci ignorovat nebo snižovat svou závislost na těchto vlastnostech (tj. těmto vlastnostem přiřadit nízkou váhu). Místo toho je model nucen hledat hlubší a méně zjevné funkce. Modely hlubokého učení obecně trpí pomalou konvergencí při trénování tohoto typu dat. Tato metoda však zlepšuje robustnost modelu a zajišťuje zobecnění. Zápornou množinu rekonstruujeme následovně. Každá pozitivní sekvence generuje jednu negativní sekvenci. Pozitivní sekvence je rozdělena do 20 podsekvenci. Poté se náhodně vybere 12 podsekcí a náhodně se nahradí. Zbývajících 8 subsekvencí je zachováno. Tento proces je znázorněn na obrázku 1. Aplikování tohoto procesu na pozitivní sadu vede k novým nepromotorovým sekvencím se zachovanými částmi z promotorových sekvencí (nezměněné subsekvence, 8 subsekvencí z 20). Tyto parametry umožňují generování negativní sady, která má 32 a 40% jejích sekvencí obsahujících konzervované části promotorových sekvencí. Bylo zjištěno, že tento poměr je optimální pro robustní prediktor promotoru, jak je vysvětleno v části 3.2.Protože konzervované části zaujímají stejné pozice v negativních sekvencích, jsou zřejmé motivy jako TATA-box a TSS nyní běžné mezi oběma sadami v poměru 32 ~ 40%. Loga sekvencí pozitivních a negativních souborů pro lidská i myší data promotoru TATA jsou uvedena na obrázcích 2, 3, v daném pořadí. Je vidět, že kladné a záporné množiny sdílejí stejné základní motivy na stejných pozicích, jako je motiv TATA na pozici -30 a –25 bp a TSS na pozici +1 bp. Školení je proto náročnější, ale výsledný model se dobře zobecňuje.
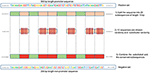
Obrázek 1. Ilustrace metody konstrukce záporné množiny. Zelená představuje náhodně konzervované subsekvence, zatímco červená představuje náhodně vybrané a substituované.

Obrázek 2. Logo sekvence v lidském promotoru TATA pro pozitivní sadu (A) i negativní sadu (B). Grafy ukazují zachování funkčních motivů mezi těmito dvěma sadami.

Obrázek 3. Logo sekvence v promotoru TATA myši pro pozitivní sadu (A) i negativní sadu (B). Pozemky ukazují zachování funkčních motivů mezi těmito dvěma sadami.
2.3. Navrhované modely
Navrhujeme model hlubokého učení, který kombinuje konvoluční vrstvy s opakujícími se vrstvami, jak je znázorněno na obrázku 4. Přijímá jednu surovou genomickou sekvenci, S = {N1, N2,…, Nl}, kde N ∈ {A, C, G, T} al je délka vstupní posloupnosti, protože vstup a výstup je skóre se skutečnou hodnotou. Vstup je jednorázově kódován a je reprezentován jako jednorozměrný vektor se čtyřmi kanály. Délka vektoru l = 300 a čtyř kanálů jsou A, C, G a T a jsou reprezentovány jako (1 0 0 0), (0 1 0 0), (0 0 1 0), (0 0 0 1 ). Abychom vybrali model s nejlepším výkonem, použili jsme metodu vyhledávání mřížky pro výběr nejlepších hyperparametrů. Vyzkoušeli jsme různé architektury, jako je samotná CNN, samotná LSTM, samotná BiLSTM, CNN kombinovaná s LSTM. Vyladěnými hyperparametry jsou počet konvolučních vrstev, velikost jádra, počet filtrů v každé vrstvě, velikost maximální sdružené vrstvy, pravděpodobnost výpadku a jednotky vrstvy Bi-LSTM.
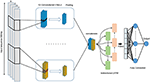
Obrázek 4. Architektura navrhovaného modelu DeePromoter.
Navrhovaný model začíná několika vrstvami konvoluce, které jsou zarovnány paralelně a pomáhají při učení důležitých motivů vstupních sekvencí s různou velikostí okna. Používáme tři konvoluční vrstvy pro non-TATA promotor s velikostí okna 27, 14 a 7 a dvě konvoluční vrstvy pro TATA promotory s velikostí okna 27, 14. Po všech konvolučních vrstvách následuje aktivační funkce ReLU (Glorot et al. , 2011), maximální sdružovací vrstva s velikostí okna 6 a odpadová vrstva s pravděpodobností 0,5. Poté jsou výstupy těchto vrstev společně zřetězeny a přiváděny do vrstvy obousměrné dlouhodobé krátkodobé paměti (BiLSTM) (Schuster a Paliwal, 1997) s 32 uzly, aby bylo možné zachytit závislosti mezi naučenými motivy z konvolučních vrstev. Naučené funkce po BiLSTM jsou zploštěny a následuje výpadek s pravděpodobností 0,5. Pak přidáme dvě plně spojené vrstvy pro klasifikaci. První má 128 uzlů a následuje ReLU a výpadek s pravděpodobností 0,5, zatímco druhá vrstva se používá pro predikci s jedním uzlem a funkcí aktivace sigmoidu. BiLSTM umožňuje, aby informace přetrvávaly a učily se dlouhodobé závislosti sekvenčních vzorků, jako je DNA a RNA. Toho je dosaženo prostřednictvím struktury LSTM, která se skládá z paměťové buňky a tří bran nazývaných brány vstupu, výstupu a zapomenutí. Tyto brány jsou odpovědné za regulaci informací v paměťové buňce. Kromě toho použití modulu LSTM zvyšuje hloubku sítě, zatímco počet požadovaných parametrů zůstává nízký. Mít hlubší síť umožňuje extrahovat složitější funkce a to je hlavním cílem našich modelů, protože negativní sada obsahuje tvrdé vzorky.
Pro konstrukci a školení navrhovaných modelů se používá rámec Keras (Chollet F. et al., 2015). Adam optimizer (Kingma and Ba, 2014) se používá k aktualizaci parametrů s rychlostí učení 0,001. Velikost dávky je nastavena na 32 a počet epoch je nastaven na 50. Včasné zastavení se použije na základě ztráty validace.
Výsledky a diskuse
3.1. Měření výkonu
V této práci používáme široce přijímané metriky hodnocení pro hodnocení výkonu navrhovaných modelů.Tyto metriky jsou přesnost, vyvolání a Matthewův korelační koeficient (MCC) a jsou definovány takto:
Kde TP je skutečně pozitivní a představuje správně identifikované promotorové sekvence, TN je skutečně negativní a představuje správně odmítnuté promotorové sekvence, FP je falešně pozitivní a představuje nesprávně identifikovaný promotorové sekvence a FN je falešně negativní a představuje nesprávně odmítnuté promotorové sekvence.
3.2. Účinek záporné množiny
Při analýze dříve publikovaných prací pro identifikaci sekvencí promotoru jsme si všimli, že výkonnost těchto prací velmi závisí na způsobu přípravy negativní datové sady. Vycházeli velmi dobře v datových sadách, které připravili, mají však vysoký falešně pozitivní poměr při hodnocení na náročnější datové sadě, která zahrnuje nepropouštěcí sekvence mající společné motivy s promotorovými sekvencemi. Například v případě datové sady promotoru TATA nebudou náhodně generované sekvence mít TATA motiv na pozici -30 a –25 bp, což zase usnadní klasifikaci. Jinými slovy, jejich klasifikátor závisel na přítomnosti TATA motivu k identifikaci promotorové sekvence a ve výsledku bylo snadné dosáhnout vysokého výkonu na souborech dat, které připravili. Jejich modely však dramaticky selhaly, když se zabývaly negativními sekvencemi, které obsahovaly motiv TATA (tvrdé příklady). Přesnost klesala, jak se zvýšila míra falešně pozitivních výsledků. Jednoduše klasifikovali tyto sekvence jako pozitivní promotorové sekvence. Podobná analýza platí pro ostatní promotorové motivy. Hlavním účelem naší práce proto není jen dosažení vysokého výkonu na konkrétní datové sadě, ale také posílení schopnosti modelu dobře generalizovat tréninkem na náročné datové sadě.
Pro další ilustraci tohoto bodu trénujeme a otestujte náš model na souborech lidských a myších TATA promotorů s různými metodami přípravy negativních sad. První experiment se provádí pomocí náhodně vzorkovaných negativních sekvencí z nekódujících oblastí genomu (tj., Podobný přístupu použitému v předchozích pracích). Je pozoruhodné, že náš navrhovaný model dosahuje téměř dokonalé přesnosti predikce (přesnost = 99%, vyvolání = 99%, Mcc = 98%) a (přesnost = 99%, vyvolání = 98%, Mcc = 97%) pro člověka i myš . Očekávají se tyto vysoké výsledky, ale otázkou je, zda si tento model může udržovat stejný výkon při hodnocení na datové sadě, která má tvrdé příklady. Odpověď na základě analýzy předchozích modelů je ne. Druhý experiment se provádí pomocí naší navrhované metody pro přípravu datové sady, jak je vysvětleno v části 2.2. Připravujeme záporné množiny, které obsahují konzervovaný TATA-box s různými procenty, jako je 12, 20, 32 a 40% a cílem je zmenšit propast mezi přesností a vyvoláním. Tím je zajištěno, že se náš model naučí složitější funkce, než aby se učil pouze přítomnost nebo nepřítomnost TATA-boxu. Jak je znázorněno na obrázcích 5A, B, model se stabilizuje v poměru 32 ~ 40% pro lidské i myší datové sady promotoru TATA.
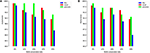
Obrázek 5. Účinek různých poměrů zachování motivu TATA v negativní sadě na výkon v případě souboru dat promotoru TATA pro člověka (A) i myš (B) .
3.3. Výsledky a srovnání
V posledních letech byla navržena řada nástrojů pro predikci promotorových oblastí (Hutchinson, 1996; Scherf et al., 2000; Reese, 2001; Umarov a Solovyev, 2017). Některé z těchto nástrojů však nejsou veřejně dostupné pro testování a některé z nich vyžadují kromě surových genomových sekvencí více informací. V této studii porovnáváme výkonnost našich navrhovaných modelů se současnou nejmodernější prací CNNProm, kterou navrhli Umarov a Solovyev (2017), jak je uvedeno v tabulce 2. Obecně platí, že navrhované modely, DeePromoter, jasně překonat CNNProm ve všech datových sadách se všemi hodnotícími metrikami. Přesněji řečeno, DeePromoter zlepšuje přesnost, vyvolání a MCC v případě lidské datové sady TATA o 0,18, 0,04 a 0,26. V případě lidské datové sady jiné než TATA zlepšuje DeePromoter přesnost o 0,39, vyvolání o 0,12 a MCC o 0,66. Podobně DeePromoter zlepšuje přesnost a MCC v případě myší datové sady TATA o 0,24, respektive 0,31. V případě myší non-TATA datové sady zlepšuje DeePromoter přesnost o 0,37, vyvolání o 0,04 a MCC o 0,65. Tyto výsledky potvrzují, že CNNProm nedokáže odmítnout negativní sekvence s TATA promotorem, proto má vysoký falešně pozitivní výsledek. Na druhou stranu jsou naše modely schopny se s těmito případy vypořádat úspěšněji a míra falešně pozitivních výsledků je nižší ve srovnání s CNNProm.

Tabulka 2. Srovnání DeePromoter se stavem metoda-the-art.
Pro další analýzy studujeme vliv střídání nukleotidů v každé poloze na výstupní skóre. Zaměřujeme se na oblast –40 a 10 bp, protože je hostitelem nejdůležitější části promotorové sekvence. Pro každou promotorovou sekvenci v testovací sadě provedeme výpočetní mutační skenování, abychom vyhodnotili účinek mutace každé báze vstupní subsekvence (150 substitucí v intervalu –40 ~ 10 bp subsekvence). To je znázorněno na obrázcích 6, 7 pro lidské datové soubory TATA a myší. Modrá barva představuje pokles výstupního skóre v důsledku mutace, zatímco červená barva představuje přírůstek skóre v důsledku mutace. Všimli jsme si, že změna nukleotidů na C nebo G v oblasti –30 a –25 bp významně snižuje výstupní skóre. Tato oblast je TATA-box, který je velmi důležitým funkčním motivem v promotorové sekvenci. Náš model tak dokáže úspěšně najít význam tohoto regionu. Ve zbytku pozic jsou nukleotidy C a G výhodnější než A a T, zejména v případě myši. To lze vysvětlit skutečností, že promotorová oblast má více nukleotidů C a G než A a T (Shi a Zhou, 2006).

Obrázek 6. Mapa výběžku oblasti –40 bp až 10 bp, která obsahuje TATA-box, v případě sekvencí lidského TATA promotoru.

Obrázek 7. Saliency map of the region –40 bp to 10 bp, which includes the TATA-box, in case of mouse TATA promotor sekvences.
Závěr
Přesná předpověď sekvencí promotoru je nezbytná pro pochopení základního mechanismu procesu genové regulace. V této práci jsme vyvinuli DeePromoter – který je založen na kombinaci konvoluční neuronové sítě a obousměrného LSTM – k predikci krátkých eukaryotických promotorových sekvencí v případě člověka a myši pro TATA i non-TATA promotor. Podstatnou součástí této práce bylo překonat problém nízké přesnosti (vysoké míry falešně pozitivních výsledků) zaznamenané v dříve vyvinutých nástrojích kvůli spoléhání se na některé zjevné rysy / motivy v sekvenci při klasifikaci promotorových a nepropagačních sekvencí. V této práci jsme se zvláště zajímali o konstrukci tvrdé záporné množiny, která pohání modely směrem k prozkoumání sekvence pro hluboké a relevantní rysy, namísto pouze rozlišení promotorových a nepropagačních sekvencí na základě existence některých funkčních motivů. Hlavní výhody používání DeePromoter spočívají v tom, že významně snižuje počet falešně pozitivních předpovědí a dosahuje vysoké přesnosti u náročných datových sad. DeePromoter překonal předchozí metodu nejen ve výkonu, ale také v překonání problému vysokých falešně pozitivních předpovědí. Předpokládá se, že tento rámec může být užitečný v aplikacích souvisejících s drogami a na akademické půdě.
Autorské příspěvky
MO a ZL připravily datový soubor, vytvořily algoritmus a provedly experiment a analýza. MO a HT připravily webový server a napsaly rukopis s podporou ZL a KC. Všichni autoři diskutovali o výsledcích a přispěli ke konečnému rukopisu.
Financování
Tento výzkum byl podpořen programem výzkumu mozku Národní výzkumné nadace (NRF) financovaným korejskou vládou ( MSIT) (č. NRF-2017M3C7A1044815).
Prohlášení o střetu zájmů
Autoři prohlašují, že výzkum byl proveden bez jakýchkoli obchodních nebo finančních vztahů, které by mohly být vykládány jako potenciální střet zájmů.
Bharanikumar, R., Premkumar, KAR a Palaniappan, A. (2018). Promoterpredict: modelování založené na sekvenci síly promotoru escherichia coli σ70 poskytuje logaritmickou závislost mezi silou promotoru a sekvencí. PeerJ 6: e5862. doi: 10,7717 / peerj.5862
PubMed Abstract | CrossRef Full Text | Google Scholar
Glorot, X., Bordes, A. a Bengio, Y. (2011). „Deep sparse rectifier neurural networks,“ ve Sborníku ze čtrnácté mezinárodní konference o umělé inteligenci a statistice (Fort Lauderdale, FL :)) 315–323.
Google Scholar
Hutchinson, G. (1996). Predikce oblastí promotorů obratlovců pomocí analýzy frekvence různých hexamerů. Bioinformatika 12, 391–398.
PubMed Abstract | Google Scholar
Kingma, DP a Ba, J. (2014). Adam: metoda pro stochastickou optimalizaci. arXiv preprint arXiv: 1412.6980.
Google Scholar
Knudsen, S. (1999). Promoter2.0: pro rozpoznávání sekvencí promotoru polii. Bioinformatika 15, 356–361.
Abstrakt PubMed | Google Scholar
Ponger, L. a Mouchiroud, D. (2002). Cpgprod: identifikace ostrovů cpg spojených s místy zahájení transkripce ve velkých genomových savčích sekvencích. Bioinformatika 18, 631–633. doi: 10,1093 / bioinformatika / 18.4.631
PubMed Abstract | CrossRef Full Text | Google Scholar
Quang, D. a Xie, X. (2016). Danq: hybridní konvoluční a rekurentní hluboká neurální síť pro kvantifikaci funkce sekvencí DNA. Nucleic Acids Res. 44, e107 – e107. doi: 10.1093 / nar / gkw226
PubMed Abstract | CrossRef Full Text | Google Scholar
Umarov, R. K. a Solovyev, V. V. (2017). Rozpoznávání prokaryotických a eukaryotických promotorů pomocí konvolučních neuronových sítí s hlubokým učením. PLoS ONE 12: e0171410. doi: 10,1371 / journal.pone.0171410
PubMed Abstract | CrossRef Full Text | Google Scholar