Grænser inden for genetik
Introduktion
Promotorer er nøgleelementerne, der hører til ikke-kodende regioner i genomet. De styrer stort set aktivering eller undertrykkelse af generne. De er placeret nær og opstrøms genets transkriptionsstartsted (TSS). Et gen’s promotorflankerende region kan indeholde mange vigtige korte DNA-elementer og motiver (5 og 15 baser lange), der tjener som genkendelsessteder for de proteiner, der tilvejebringer korrekt initiering og regulering af transkription af downstream-genet (Juven-Gershon et al., 2008). Indledningen af gentranskription er det mest grundlæggende trin i reguleringen af genekspression. Promotorkerne er en minimal strækning af DNA-sekvens, som kegler TSS og er tilstrækkelig til direkte at initiere transkriptionen. Længden af kernepromotor varierer typisk mellem 60 og 120 basepar (bp).
TATA-boksen er en promotorsekvens, der angiver for andre molekyler, hvor transkription begynder. Den blev navngivet “TATA-box”, da dens sekvens er kendetegnet ved gentagelse af T- og A-basepar (TATAAA) (Baker et al., 2003). Langt de fleste undersøgelser af TATA-boxen er blevet udført på mennesker, gær, og Drosophila genomer er der imidlertid fundet lignende elementer i andre arter såsom arkæer og gamle eukaryoter (Smale og Kadonaga, 2003). I humane tilfælde har 24% af gener promotorregioner indeholdende TATA-box (Yang et al., 2007 I eukaryoter er TATA-box placeret ved ~ 25 bp opstrøms for TSS (Xu et al., 2016). Den er i stand til at definere transkriptionsretningen og indikerer også den DNA-streng, der skal læses. Proteiner kaldet transkriptionsfaktorer binder til flere ikke-kodende regioner inklusive TATA-box og rekruttere et enzym kaldet RNA-polymerase, som syntetiserer RNA fra DNA.
På grund af promotorernes vigtige rolle i gentranskription bliver nøjagtig forudsigelse af promotorsteder et krævet trin i genekspression, fortolkning af mønstre og opbygning og forståelse funktionaliteten af genetiske reguleringsnetværk. Der var forskellige biologiske eksperimenter til identifikation af promotorer såsom mutationsanalyse (Matsumine et al., 1998) og immunpræcipiteringsassays (Kim et al., 2004; Dahl og Collas, 2008). Disse metoder var imidlertid både dyre og tidskrævende. For nylig med udviklingen af næste generations sekventering (NGS) (Behjati og Tarpey, 2013) er flere gener fra forskellige organismer blevet sekventeret, og deres genelementer er blevet udforsket beregningsmæssigt (Zhang et al., 2011). På den anden side har innovationen af NGS-teknologi resulteret i et dramatisk fald i omkostningerne ved hele genom-sekventeringen, således at der er flere sekventeringsdata tilgængelige. Datatilgængeligheden tiltrækker forskere til at udvikle beregningsmodeller til promotor forudsigelsesopgave. Det er dog stadig en ufuldstændig opgave, og der er ingen effektiv software, der nøjagtigt kan forudsige promotorer.
Promotorprædiktorer kan kategoriseres baseret på den anvendte tilgang i tre grupper, nemlig signalbaseret tilgang, indholdsbaseret tilgang og den GpG-baserede tilgang. Signalbaserede forudsigere fokuserer på promotorelementer relateret til RNA-polymerase-bindingssted og ignorerer ikke-elementdelene af sekvensen. Som et resultat var forudsigelsesnøjagtigheden svag og ikke tilfredsstillende. Eksempler på signalbaserede forudsigere inkluderer: PromoterScan (Prestridge, 1995), der anvendte de ekstraherede træk i TATA-boksen og en vægtet matrix af transkriptionsfaktorbindingssites med en lineær diskriminator til at klassificere promotorsekvenser fra ikke-promotorsekvenser; Promoter2.0 (Knudsen, 1999), som ekstraherede funktionerne fra forskellige kasser såsom TATA-Box, CAAT-Box og GC-Box og sendte dem til kunstige neurale netværk (ANN) til klassificering; NNPP2.1 (Reese, 2001), der anvendte initiatorelement (Inr) og TATA-Box til ekstraktion af funktioner og et tidsforsinket neuralt netværk til klassificering, og Down and Hubbard (2002), der brugte TATA-Box og anvendte en relevansvektormaskiner (RVM) som klassifikator. Indholdsbaserede forudsigere var afhængige af at tælle frekvensen af k-mer ved at køre et vindue med k-længde på tværs af sekvensen. Imidlertid ignorerer disse metoder den rumlige information om baseparret i sekvenserne. Eksempler på indholdsbaserede forudsigere inkluderer: PromFind (Hutchinson, 1996), der anvendte k-mer-frekvensen til at udføre forudsigelsen af hexamerpromotoren; PromoterInspector (Scherf et al., 2000), som identificerede regionerne indeholdende promotorer baseret på en fælles genomisk kontekst af polymerase II-promotorer ved at scanne efter specifikke træk defineret som motiver med variabel længde; MCPromoter1.1 (Ohler et al., 1999), der anvendte en enkelt interpoleret Markov-kæde (IMC) af 5. orden til at forudsige promotorsekvenser.Endelig udnyttede GpG-baserede forudsigere placeringen af GpG-øer som promotorregion eller den første exonregion i de humane gener indeholder normalt GpG-øer (Ioshikhes og Zhang, 2000; Davuluri et al., 2001; Lander et al., 2001; Ponger og Mouchiroud, 2002). Imidlertid indeholder kun 60% af promotorerne GpG-øer, hvorfor forudsigelsesnøjagtigheden af denne type forudsigere aldrig oversteg 60%.
For nylig er sekvensbaserede fremgangsmåder blevet anvendt til promotor forudsigelse. Yang et al. (2017) anvendte forskellige funktionsekstraktionsstrategier til at indfange de mest relevante sekvensoplysninger for at forudsige interaktioner mellem forstærker og promotor. Lin et al. (2017) foreslog en sekvensbaseret forudsigelse, navngivet “iPro70-PseZNC”, til sigma70-promotors identifikation i prokaryoten. Ligeledes Bharanikumar et al. (2018) foreslog PromoterPredict for at forudsige styrken af Escherichia coli-promotorer baseret på en dynamisk multipel regressionsmetode, hvor sekvenserne blev repræsenteret som positionsvægtmatricer (PWM). Kanhere og Bansal (2005) udnyttede forskellene i DNA-sekvensstabilitet mellem promotor- og ikke-promotorsekvenserne for at skelne mellem dem. Xiao et al. (2018) introducerede en to-lags forudsigelse kaldet iPSW (2L) -PseKNC til promotorsekvensidentifikation samt styrken af promotorerne ved at udvinde hybridfunktioner fra sekvenserne.
Alle de førnævnte forudsigere kræver domæne- viden for at håndarbejde funktionerne. På den anden side muliggør dyb læringsbaserede tilgange mulighed for at opbygge mere effektive modeller ved hjælp af rådata (DNA / RNA-sekvenser) direkte. Dybt konvolutionsneuralt netværk opnåede avancerede resultater i udfordrende opgaver såsom behandling af billede, video, lyd og tale (Krizhevsky et al., 2012; LeCun et al., 2015; Schmidhuber, 2015; Szegedy et al. , 2015). Derudover blev det med succes anvendt i biologiske problemer som DeepBind (Alipanahi et al., 2015), DeepCpG (Angermueller et al., 2017), valg af forgreningspunkt (Nazari et al., 2018), alternative splejningssteder forudsigelse (Oubounyt et al., 2018), 2 “-Omethyleringssteder forudsigelse (Tahir et al., 2018), DNA-sekvenskvantificering (Quang og Xie, 2016), humant protein subcellulær lokalisering (Wei et al., 2018) osv. Endvidere CNN fik for nylig betydelig opmærksomhed i promotorgenkendelsesopgaven. Meget for nylig introducerede Umarov og Solovyev (2017) CNNprom til diskrimination af korte promotorsekvenser, denne CNN-baserede arkitektur opnåede høje resultater i klassificeringen af promotorsekvenser og ikke-promotorsekvenser. Derefter blev denne model forbedret af Qian et al. (2018), hvor forfatterne brugte SVM-klassifikator (support vector machine) til at inspicere de vigtigste promotorsekvenselementer. Dernæst blev de mest indflydelsesrige elementer holdt ukomprimeret, mens de mindre vigtige komprimeres. Denne proces resulterede i bedre ydeevne. For nylig blev lang promotoridentifikationsmodel foreslået af Umarov et al. (2019) hvor forfatterne fokuserede på identifikationen af TSS-position.
I alle de ovennævnte værker blev det negative sæt ekstraheret fra ikke-promotorregioner i genomet. At vide, at promotorsekvenserne udelukkende er rige på specifikke funktionelle elementer såsom TATA-box, som er placeret ved –30 ~ –25 bp, GC-Box, som er placeret ved –110 ~ –80 bp, CAAT-Box, som er placeret ved – 80 ~ –70 bp osv. Dette resulterer i høj klassifikationsnøjagtighed på grund af enorm forskel mellem de positive og negative prøver med hensyn til sekvensstruktur. Derudover bliver klassificeringsopgaven ubesværet at opnå, for eksempel vil CNN-modeller bare stole på tilstedeværelsen eller fraværet af nogle motiver i deres specifikke positioner for at træffe beslutningen om sekvenstypen. Således har disse modeller meget lav præcision / følsomhed (høj falsk positiv), når de testes på genomiske sekvenser, der har promotormotiver, men de er ikke promotorsekvenser. Det er velkendt, at der er flere TATAAA-motiver i genomet end dem, der hører til promotorregionerne. For eksempel indeholder DNA-sekvensen af det humane kromosom 1 alene, ftp://ftp.ensembl.org/pub/release-57/fasta/homo_sapiens/dna/, 151 656 TATAAA-motiver. Det er mere end det omtrentlige maksimale antal gener i det samlede humane genom. Som en illustration af dette problem bemærker vi, at når vi tester disse modeller på ikke-promotorsekvenser, der har TATA-box, forklassificerer de de fleste af disse sekvenser. Derfor, for at generere en robust klassificering, skal det negative sæt vælges omhyggeligt, da det bestemmer de funktioner, der skal bruges af klassificeren for at diskriminere klasserne. Vigtigheden af denne idé er blevet demonstreret i tidligere værker som (Wei et al., 2014). I dette arbejde behandler vi hovedsageligt dette emne og foreslår en tilgang, der integrerer nogle af de positive klassefunktionelle motiver i den negative klasse for at reducere modelens afhængighed af disse motiver.Vi bruger en CNN kombineret med LSTM-model til at analysere sekvenskarakteristika for humane og mus TATA og ikke-TATA eukaryote promotorer og opbygge beregningsmodeller, der nøjagtigt kan skelne korte promotorsekvenser fra ikke-promotors.
Materialer og metoder
2.1. Datasæt
Datasættene, der bruges til at træne og teste den foreslåede promotorprædiktor, er samlet fra menneske og mus. De indeholder to karakteristiske klasser af promotorerne, nemlig TATA-promotorer (dvs. sekvenserne, der indeholder TATA-box) og ikke-TATA-promotorer. Disse datasæt blev bygget fra Eukaryotic Promoter Database (EPDnew) (Dreos et al., 2012). EPDnew er et nyt afsnit under det velkendte EPD-datasæt (Périer et al., 2000), der er kommenteret en ikke-redundant samling af eukaryote POL II-promotorer, hvor transkriptionsstartsted er blevet bestemt eksperimentelt. Det giver promotorer af høj kvalitet sammenlignet med ENSEMBL-promotorsamling (Dreos et al., 2012), og det er offentligt tilgængeligt på https://epd.epfl.ch//index.php. Vi downloadede genomiske TATA- og ikke-TATA-promotorsekvenser for hver organisme fra EPDnew. Denne operation resulterede i opnåelse af fire promotordatasæt, nemlig: Human-TATA, Human-non-TATA, Mouse-TATA og Mouse-non-TATA. For hvert af disse datasæt konstrueres et negativt sæt (ikke-promotorsekvenser) med den samme størrelse som det positive baseret på den foreslåede tilgang som beskrevet i det følgende afsnit. Detaljerne om antallet af promotorsekvenser for hver organisme er givet i tabel 1. Alle sekvenser har en længde på 300 bp og blev ekstraheret fra -249 ~ + 50 bp (+1 henviser til TSS-position). Som kvalitetskontrol brugte vi 5 gange krydsvalidering til at vurdere den foreslåede model. I dette tilfælde bruges 3-fold til træning, 1-fold bruges til validering, og den resterende fold bruges til test. Således trænes den foreslåede model 5 gange, og den samlede præstation for 5 gange beregnes.

Tabel 1. Statistik over de fire datasæt, der er brugt i denne undersøgelse.
2.2. Negativ datasætkonstruktion
For at træne en model, der nøjagtigt kan udføre promotor- og ikke-promotorsekvensklassificering, er vi nødt til at vælge det negative sæt (ikke-promotorsekvenser) omhyggeligt. Dette punkt er afgørende for at gøre en model i stand til at generalisere godt og derfor være i stand til at opretholde sin præcision, når den vurderes på mere udfordrende datasæt. Tidligere værker, såsom (Qian et al., 2018), konstruerede negativt sæt ved tilfældigt at vælge fragmenter fra genom-ikke-promotorregioner. Det er klart, at denne tilgang ikke er helt rimelig, for hvis der ikke er noget skæringspunkt mellem positive og negative sæt. Således vil modellen let finde grundlæggende funktioner til at adskille de to klasser. For eksempel kan TATA-motiv findes i alle positive sekvenser i en bestemt position (normalt 28 bp opstrøms for TSS, mellem –30 og –25 pb i vores datasæt). Derfor vil oprettelse af et negativt sæt tilfældigt, der ikke indeholder dette motiv, producere høj ydeevne i dette datasæt. Modellen mislykkes imidlertid med at klassificere negative sekvenser, der har TATA-motiv som promotorer. Kort fortalt er den største fejl i denne tilgang, at når man træner en dyb læringsmodel, lærer den kun at diskriminere de positive og negative klasser baseret på tilstedeværelsen eller fraværet af nogle enkle funktioner på specifikke positioner, hvilket gør disse modeller umulige. I dette arbejde tilstræber vi at løse dette problem ved at etablere en alternativ metode til at udlede det negative sæt fra det positive.
Vores metode er baseret på det faktum, at når funktionerne er fælles mellem det negative og det positiv klasse, når modellen træffer beslutningen, har den tendens til at ignorere eller reducere dens afhængighed af disse funktioner (dvs. tildele lave vægte til disse funktioner). I stedet er modellen tvunget til at søge efter dybere og mindre åbenlyse funktioner. Deep learning-modeller lider generelt under langsom konvergens under træning i denne type data. Denne metode forbedrer dog robustheden i modellen og sikrer generalisering. Vi rekonstruerer det negative sæt som følger. Hver positive sekvens genererer en negativ sekvens. Den positive sekvens er opdelt i 20 sekvenser. Derefter vælges 12 efterfølgende tilfældigt og erstattes tilfældigt. De resterende otte sekvenser bevares. Denne proces er illustreret i figur 1. Anvendelse af denne proces på det positive sæt resulterer i nye ikke-promotorsekvenser med konserverede dele fra promotorsekvenser (de uændrede sekvenser, 8 sekvenser ud af 20). Disse parametre muliggør generering af et negativt sæt, der har 32 og 40% af dets sekvenser, der indeholder konserverede dele af promotorsekvenser. Dette forhold viser sig at være optimalt til at have en robust promotorprædiktor som forklaret i afsnit 3.2.Fordi de konserverede dele indtager de samme positioner i de negative sekvenser, er de åbenlyse motiver som TATA-box og TSS nu almindelige mellem de to sæt med et forhold på 32 ~ 40%. Sekvenslogoerne for de positive og negative sæt for både human og mus TATA-promotordata er vist i henholdsvis figur 2, 3. Det kan ses, at de positive og de negative sæt deler de samme grundlæggende motiver i de samme positioner som TATA-motivet ved positionen -30 og –25 bp og TSS ved positionen +1 bp. Derfor er træningen mere udfordrende, men den resulterede model generaliserer godt.
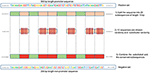
Figur 1. Illustration af den negative sætkonstruktionsmetode. Grøn repræsenterer de tilfældigt konserverede sekvenser, mens rød repræsenterer tilfældigt valgte og substituerede.

Figur 2. Sekvenslogoet i human TATA-promotor for både positivt sæt (A) og negativt sæt (B). Diagrammerne viser bevarelsen af de funktionelle motiver mellem de to sæt.

Figur 3. Sekvenslogoet i mus TATA-promotor for både positivt sæt (A) og negativt sæt (B). Plottene viser bevarelsen af de funktionelle motiver mellem de to sæt.
2.3. De foreslåede modeller
Vi foreslår en dyb læringsmodel, der kombinerer sammenfoldningslag med tilbagevendende lag som vist i figur 4. Den accepterer en enkelt rå genomisk sekvens, S = {N1, N2,…, Nl} hvor N ∈ {A, C, G, T} og l er længden af indgangssekvensen, da input og output udsendes en reel værdi. Indgangen er kodet én-hot og repræsenteret som en endimensionel vektor med fire kanaler. Længden af vektoren l = 300 og de fire kanaler er A, C, G og T og repræsenteret som (1 0 0 0), (0 1 0 0), (0 0 1 0), (0 0 0 1 ), henholdsvis. For at vælge den bedst mulige model har vi brugt gittersøgemetode til at vælge de bedste hyperparametre. Vi har prøvet forskellige arkitekturer såsom CNN alene, LSTM alene, BiLSTM alene, CNN kombineret med LSTM. De indstillede hyperparametre er antallet af sammenviklingslag, kernestørrelse, antal filtre i hvert lag, størrelsen på det maksimale poollag, sandsynligheden for frafald og enhederne i Bi-LSTM-laget.
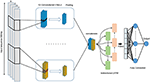
Figur 4. Arkitekturen for den foreslåede DeePromoter-model.
Den foreslåede model starter med flere foldningslag, der er justeret parallelt og hjælper med at lære de vigtige motiver i input-sekvenserne med forskellige vinduesstørrelser. Vi bruger tre sammenblandingslag til ikke-TATA-promotor med vinduesstørrelser på 27, 14 og 7 og to sammenblandingslag til TATA-promotorer med vinduesstørrelser på 27, 14. Alle sammenblandingslag efterfølges af ReLU-aktiveringsfunktion (Glorot et al. , 2011), et maks. Poolinglag med en vinduesstørrelse på 6 og et dropout-lag med en sandsynlighed på 0,5. Derefter sammenkædes outputene fra disse lag og føres ind i et tovejs kortvarig hukommelse (BiLSTM) (Schuster og Paliwal, 1997) med 32 knudepunkter for at fange afhængighederne mellem de indlærte motiver fra konvolutionslagene. De lærte funktioner efter BiLSTM er fladtrykt og efterfulgt af frafald med en sandsynlighed på 0,5. Derefter tilføjer vi to fuldt forbundne lag til klassificering. Den første har 128 noder og efterfulgt af ReLU og dropout med en sandsynlighed på 0,5, mens det andet lag bruges til forudsigelse med en node og sigmoid aktiveringsfunktion. BiLSTM tillader, at informationen fortsætter og lærer langsigtede afhængigheder af sekventielle prøver såsom DNA og RNA. Dette opnås gennem LSTM-strukturen, der er sammensat af en hukommelsescelle og tre porte kaldet input-, output- og glemeporte. Disse porte er ansvarlige for at regulere informationen i hukommelsescellen. Derudover øger brugen af LSTM-modulet netværksdybden, mens antallet af de krævede parametre forbliver lavt. At have et dybere netværk muliggør ekstrahering af mere komplekse funktioner, og dette er hovedformålet med vores modeller, da det negative sæt indeholder hårde prøver.
Keras-rammen bruges til at konstruere og træne de foreslåede modeller (Chollet F. et. al., 2015). Adam optimizer (Kingma og Ba, 2014) bruges til at opdatere parametrene med en læringshastighed på 0,001. Batchstørrelsen er indstillet til 32, og antallet af epoker er indstillet til 50. Tidlig standsning anvendes baseret på valideringstab.
Resultater og diskussion
3.1. Ydelsesforanstaltninger
I dette arbejde bruger vi de bredt vedtagne evalueringsmålinger til at evaluere præstationen for de foreslåede modeller.Disse metrics er præcision, tilbagekaldelse og Matthew korrelationskoefficient (MCC), og de defineres som følger:
Hvor TP er sandt positiv og repræsenterer korrekt identificerede promotorsekvenser, er TN sandt negativ og repræsenterer korrekt afviste promotorsekvenser, FP er falsk positiv og repræsenterer forkert identificeret promotorsekvenser, og FN er falsk negativ og repræsenterer forkert afviste promotorsekvenser.
3.2. Effekt af det negative sæt
Når vi analyserede de tidligere offentliggjorte værker til identifikation af promotorsekvenser, bemærkede vi, at udførelsen af disse værker i høj grad afhænger af, hvordan man forbereder det negative datasæt. De klarede sig meget godt på de datasæt, de har udarbejdet, men de har et højt falsk positivt forhold, når de vurderes på et mere udfordrende datasæt, der inkluderer ikke-prompter-sekvenser med fælles motiver med promotorsekvenser. For eksempel i tilfælde af TATA-promotordatasættet vil de tilfældigt genererede sekvenser ikke have TATA-motiv ved positionen -30 og –25 bp, hvilket igen gør opgaven med klassificering lettere. Med andre ord afhængede deres klassifikator af tilstedeværelsen af TATA-motiv for at identificere promotorsekvensen, og som et resultat var det let at opnå høj ydeevne på de datasæt, de har forberedt. Imidlertid mislykkedes deres modeller dramatisk, når de beskæftiger sig med negative sekvenser, der indeholdt TATA-motiv (hårde eksempler). Præcisionen faldt, efterhånden som den falske positive sats steg. Simpelthen klassificerede de disse sekvenser som positive promotorsekvenser. En lignende analyse er gyldig for de andre promotormotiver. Derfor er hovedformålet med vores arbejde ikke kun at opnå høj ydeevne på et specifikt datasæt, men også at forbedre modelevnen til at generalisere godt ved at træne i et udfordrende datasæt.
For mere at illustrere dette punkt træner vi og test vores model på TATA-promotors datasæt til mennesker og mus med forskellige metoder til forberedelse af negative sæt. Det første eksperiment udføres ved anvendelse af tilfældigt samplede negative sekvenser fra ikke-kodende regioner i genomet (dvs. svarer til fremgangsmåden anvendt i de tidligere værker). Bemærkelsesværdigt opnår vores foreslåede model næsten perfekt forudsigelsesnøjagtighed (præcision = 99%, tilbagekaldelse = 99%, Mcc = 98%) og (præcision = 99%, tilbagekaldelse = 98%, Mcc = 97%) for henholdsvis både menneske og mus . Disse høje resultater forventes, men spørgsmålet er, om denne model kan opretholde den samme ydeevne, når den vurderes på et datasæt, der har hårde eksempler. Svaret, baseret på analyse af de tidligere modeller, er nej. Det andet eksperiment udføres ved hjælp af vores foreslåede metode til at forberede datasættet som forklaret i afsnit 2.2. Vi forbereder de negative sæt, der indeholder konserveret TATA-boks med forskellige procenter som 12, 20, 32 og 40%, og målet er at reducere afstanden mellem præcision og tilbagekaldelse. Dette sikrer, at vores model lærer mere komplekse funktioner i stedet for kun at lære tilstedeværelsen eller fraværet af TATA-box. Som vist i figur 5A stabiliserer B modellen sig i forholdet 32 ~ 40% for både TATA-promotors datasæt for mennesker og mus.
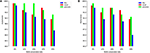
Figur 5. Virkningen af forskellige konserveringsforhold for TATA-motiv i det negative sæt på ydeevnen i tilfælde af TATA-promotordatasæt for både human (A) og mus (B) .
3.3. Resultater og sammenligning
I løbet af de sidste år er der blevet foreslået masser af forudsigelsesværktøjer til promotorregionen (Hutchinson, 1996; Scherf et al., 2000; Reese, 2001; Umarov og Solovyev, 2017). Nogle af disse værktøjer er imidlertid ikke offentligt tilgængelige til test, og nogle af dem kræver mere information udover de rå genomiske sekvenser. I denne undersøgelse sammenligner vi ydeevnen for vores foreslåede modeller med det aktuelle avancerede arbejde, CNNProm, som blev foreslået af Umarov og Solovyev (2017) som vist i tabel 2. Generelt er de foreslåede modeller, DeePromoter, klart bedre end CNNProm i alle datasæt med alle evalueringsmålinger. Mere specifikt forbedrer DeePromoter præcisionen, tilbagekaldelsen og MCC i tilfælde af humant TATA-datasæt med henholdsvis 0,18, 0,04 og 0,26. I tilfælde af humant ikke-TATA-datasæt forbedrer DeePromoter præcisionen med 0,39, tilbagekaldelsen med 0,12 og MCC med 0,66. Tilsvarende forbedrer DeePromoter præcisionen og MCC i tilfælde af mus TATA-datasæt med henholdsvis 0,24 og 0,31. I tilfælde af mus, der ikke er TATA-datasæt, forbedrer DeePromoter præcisionen med 0,37, tilbagekaldelsen med 0,04 og MCC med 0,65. Disse resultater bekræfter, at CNNProm ikke afviser negative sekvenser med TATA-promotor, derfor har den høj falsk positiv. På den anden side er vores modeller i stand til at håndtere disse sager mere vellykket, og falsk positiv sats er lavere sammenlignet med CNNProm.

Tabel 2. Sammenligning af DeePromoter med status for -the-art-metoden.
For yderligere analyser studerer vi effekten af skiftende nukleotider i hver position på outputscoren. Vi fokuserer på regionen –40 og 10 bp, da den er vært for den vigtigste del af promotorsekvensen. For hver promotorsekvens i testsættet udfører vi beregningsmutationsscanning for at evaluere effekten af at mutere hver base af indgangssekvensen (150 substitutioner på intervallet –40 ~ 10 bp efterfølgende). Dette er illustreret i figur 6, 7 for henholdsvis humane TATA-datasæt og musesæt. Blå farve repræsenterer et fald i output score på grund af mutation, mens den røde farve repræsenterer stigningen i score på grund af mutation. Vi bemærker, at ændring af nukleotiderne til C eller G i regionen –30 og –25 bp reducerer output score markant. Denne region er TATA-box, som er et meget vigtigt funktionelt motiv i promotorsekvensen. Således er vores model med succes i stand til at finde vigtigheden af denne region. I de øvrige positioner foretrækkes C- og G-nukleotider mere end A og T, især i tilfælde af musen. Dette kan forklares ved, at promotorregionen har flere C- og G-nukleotider end A og T (Shi og Zhou, 2006).

Figur 6. Fremhævelseskortet for regionen –40 bp til 10 bp, som inkluderer TATA-boksen, i tilfælde af humane TATA-promotorsekvenser.

Figur 7. Fremhævelseskortet over regionen –40 bp til 10 bp, som inkluderer TATA-boksen, i tilfælde af mus TATA-promotorsekvenser.
Konklusion
Nøjagtig forudsigelse af promotorsekvenser er vigtig for at forstå den underliggende mekanisme i genreguleringsprocessen. I dette arbejde udviklede vi DeePromoter -som er baseret på en kombination af neuralt netværk med to-vejs-opløsning og LSTM- til at forudsige de korte eukaryote promotorsekvenser i tilfælde af menneske og mus for både TATA og ikke-TATA-promotor. Den væsentlige komponent i dette arbejde var at overvinde spørgsmålet om lav præcision (høj falsk positiv hastighed), der blev bemærket i de tidligere udviklede værktøjer på grund af afhængigheden af nogle indlysende træk / motiver i sekvensen ved klassificering af promotor- og ikke-promotorsekvenser. I dette arbejde var vi især interesserede i at konstruere et hårdt negativt sæt, der driver modellerne mod at udforske sekvensen for dybe og relevante funktioner i stedet for kun at skelne mellem promotor- og ikke-promotorsekvenser baseret på eksistensen af nogle funktionelle motiver. De største fordele ved at bruge DeePromoter er, at det reducerer antallet af falske positive forudsigelser betydeligt, samtidig med at der opnås høj nøjagtighed på udfordrende datasæt. DeePromoter overgik den tidligere metode ikke kun i forestillingen, men også i at overvinde spørgsmålet om høje falske positive forudsigelser. Det forventes, at denne ramme kan være nyttig i narkotikarelaterede applikationer og den akademiske verden.
Forfatterbidrag
MO og ZL forberedte datasættet, udtænkte algoritmen og gennemførte eksperimentet og analyse. MO og HT forberedte webserveren og skrev manuskriptet med støtte fra ZL og KC. Alle forfattere diskuterede resultaterne og bidrog til det endelige manuskript.
Finansiering
Denne forskning blev støttet af hjerneforskningsprogrammet fra National Research Foundation (NRF) finansieret af den koreanske regering ( MSIT) (Nr. NRF-2017M3C7A1044815).
Erklæring om interessekonflikt
Forfatterne erklærer, at forskningen blev udført i fravær af nogen kommercielle eller økonomiske forhold, der kunne fortolkes som en potentiel interessekonflikt.
Bharanikumar, R., Premkumar, KAR og Palaniappan, A. (2018). Promoterforudsigelse: sekvensbaseret modellering af escherichia coli σ70 promotorstyrke giver logaritmisk afhængighed mellem promotorstyrke og sekvens. PeerJ 6: e5862. doi: 10.7717 / peerj.5862
PubMed Abstract | CrossRef fuldtekst | Google Scholar
Glorot, X., Bordes, A. og Bengio, Y. (2011). “Deep sparse rectifier neurale netværk,” i Proceedings of the Fjerenthenth International Conference on Artificial Intelligence and Statistics, (Fort Lauderdale, FL:) 315–323.
Google Scholar
Hutchinson, G. (1996). Forudsigelsen af hvirveldyrpromotorregioner ved anvendelse af differentiel hexamerfrekvensanalyse. Bioinformatik 12, 391–398.
PubMed Abstract | Google Scholar
Kingma, DP og Ba, J. (2014). Adam: en metode til stokastisk optimering. arXiv preprint arXiv: 1412.6980.
Google Scholar
Knudsen, S. (1999). Promoter2. 0: til anerkendelse af polii-promotorsekvenser. Bioinformatik 15, 356-361.
PubMed Abstract | Google Scholar
Ponger, L. og Mouchiroud, D. (2002). Cpgprod: identificering af cpg-øer associeret med transkriptionsstartsteder i store genomiske pattedyrsekvenser. Bioinformatik 18, 631-633. doi: 10.1093 / bioinformatics / 18.4.631
PubMed Abstract | CrossRef fuldtekst | Google Scholar
Quang, D. og Xie, X. (2016). Danq: et hybrid konvolutions- og tilbagevendende dybt neuralt netværk til kvantificering af funktionen af dna-sekvenser. Nucleic Acids Res. 44, e107 – e107. doi: 10.1093 / nar / gkw226
PubMed Abstract | CrossRef fuldtekst | Google Scholar
Umarov, R. K. og Solovyev, V. V. (2017). Anerkendelse af prokaryote og eukaryote promotorer ved hjælp af nedbrydning af neurale neurale netværk. PLoS ONE 12: e0171410. doi: 10.1371 / journal.pone.0171410
PubMed Abstract | CrossRef fuldtekst | Google Scholar