Fronteras en genética
Introducción
Los promotores son los elementos clave que pertenecen a regiones no codificantes del genoma. Controlan en gran medida la activación o represión de los genes. Están ubicados cerca y corriente arriba del sitio de inicio de la transcripción del gen (TSS). La región flanqueante del promotor de un gen puede contener muchos elementos y motivos cruciales de ADN corto (5 y 15 bases de largo) que sirven como sitios de reconocimiento para las proteínas que proporcionan iniciación y regulación adecuadas de la transcripción del gen aguas abajo (Juven-Gershon et al., 2008). El inicio de la transcripción de genes es el paso más fundamental en la regulación de la expresión de genes. El núcleo promotor es un tramo mínimo de la secuencia de ADN que combina TSS y es suficiente para iniciar directamente la transcripción. La longitud del promotor central suele oscilar entre 60 y 120 pares de bases (pb).
La caja TATA es una subsecuencia del promotor que indica a otras moléculas dónde comienza la transcripción. Se le denominó «caja TATA» porque su secuencia se caracteriza por pares de bases T y A repetidos (TATAAA) (Baker et al., 2003). La gran mayoría de los estudios sobre la caja TATA se han realizado en humanos, levaduras, y genomas de Drosophila, sin embargo, se han encontrado elementos similares en otras especies como arqueas y eucariotas ancestrales (Smale y Kadonaga, 2003). En el caso humano, el 24% de los genes tienen regiones promotoras que contienen TATA-box (Yang et al., 2007 ). En eucariotas, TATA-box se encuentra a ~ 25 pb corriente arriba del TSS (Xu et al., 2016). Es capaz de definir la dirección de la transcripción y también indica la cadena de ADN que se debe leer. Proteínas llamadas factores de transcripción se unen a varias regiones no codificantes, incluida la caja TATA, y reclutan una enzima llamada ARN polimerasa, que sintetiza ARN a partir del ADN.
Debido al importante papel de los promotores en la transcripción de genes, la predicción precisa de los sitios del promotor se vuelve un paso necesario en la expresión génica, la interpretación de patrones y la construcción y comprensión la funcionalidad de las redes reguladoras genéticas. Se realizaron diferentes experimentos biológicos para la identificación de promotores como el análisis mutacional (Matsumine et al., 1998) y los ensayos de inmunoprecipitación (Kim et al., 2004; Dahl y Collas, 2008). Sin embargo, estos métodos eran costosos y consumían mucho tiempo. Recientemente, con el desarrollo de la secuenciación de próxima generación (NGS) (Behjati y Tarpey, 2013) se han secuenciado más genes de diferentes organismos y se han explorado computacionalmente sus elementos genéticos (Zhang et al., 2011). Por otro lado, la innovación de la tecnología NGS ha resultado en una caída dramática del costo de la secuenciación del genoma completo, por lo que se dispone de más datos de secuenciación. La disponibilidad de datos atrae a los investigadores a desarrollar modelos computacionales para la tarea de predicción de promotores. Sin embargo, todavía es una tarea incompleta y no existe un software eficiente que pueda predecir con precisión los promotores.
Los predictores de promotores pueden clasificarse según el enfoque utilizado en tres grupos: enfoque basado en señales, enfoque basado en contenido y el enfoque basado en GpG. Los predictores basados en señales se centran en los elementos promotores relacionados con el sitio de unión de la ARN polimerasa e ignoran las porciones que no son elementos de la secuencia. Como resultado, la precisión de la predicción fue débil y no satisfactoria. Ejemplos de predictores basados en señales incluyen: PromoterScan (Prestridge, 1995) que utilizó las características extraídas de la caja TATA y una matriz ponderada de sitios de unión de factores de transcripción con un discriminador lineal para clasificar las secuencias promotoras de las no promotoras; Promoter2.0 (Knudsen, 1999) que extrajo las características de diferentes cajas como TATA-Box, CAAT-Box y GC-Box y las pasó a redes neuronales artificiales (ANN) para su clasificación; NNPP2.1 (Reese, 2001) que utilizó el elemento iniciador (Inr) y TATA-Box para la extracción de características y una red neuronal de retardo de tiempo para la clasificación, y Down y Hubbard (2002) que utilizaron TATA-Box y utilizaron máquinas vectoriales de relevancia (RVM) como clasificador. Los predictores basados en contenido se basaban en contar la frecuencia de k-mer ejecutando una ventana de longitud k en la secuencia. Sin embargo, estos métodos ignoran la información espacial de los pares de bases en las secuencias. Ejemplos de predictores basados en contenido incluyen: PromFind (Hutchinson, 1996) que usó la frecuencia k-mer para realizar la predicción del promotor hexámero; PromoterInspector (Scherf et al., 2000) que identificó las regiones que contienen promotores basándose en un contexto genómico común de promotores de la polimerasa II mediante la exploración de características específicas definidas como motivos de longitud variable; MCPromoter1.1 (Ohler et al., 1999) que utilizó una única cadena de Markov interpolada (IMC) de quinto orden para predecir secuencias promotoras.Finalmente, los predictores basados en GpG utilizaron la ubicación de las islas GpG como la región promotora o la primera región del exón en los genes humanos generalmente contiene islas GpG (Ioshikhes y Zhang, 2000; Davuluri et al., 2001; Lander et al., 2001; Ponger y Mouchiroud, 2002). Sin embargo, solo el 60% de los promotores contienen islas GpG, por lo que la precisión de predicción de este tipo de predictores nunca superó el 60%.
Recientemente, se han utilizado enfoques basados en secuencias para la predicción de promotores. Yang y col. (2017) utilizaron diferentes estrategias de extracción de características para capturar la información de secuencia más relevante con el fin de predecir las interacciones potenciador-promotor. Lin y col. (2017) propuso un predictor basado en secuencia, llamado «iPro70-PseZNC», para la identificación del promotor sigma70 en el procariota. Asimismo, Bharanikumar et al. (2018) propuso PromoterPredict para predecir la fuerza de los promotores de Escherichia coli en base a un enfoque de regresión múltiple dinámica donde las secuencias se representaron como matrices de peso de posición (PWM). Kanhere y Bansal (2005) utilizaron las diferencias en la estabilidad de la secuencia de ADN entre las secuencias promotoras y no promotoras para distinguirlas. Xiao y col. (2018) introdujeron un predictor de dos capas llamado iPSW (2L) -PseKNC para la identificación de secuencias promotoras, así como la fuerza de los promotores mediante la extracción de características híbridas de las secuencias.
Todos los predictores antes mencionados requieren dominio- conocimiento para elaborar a mano las características. Por otro lado, los enfoques basados en el aprendizaje profundo permiten construir modelos más eficientes utilizando datos en bruto (secuencias de ADN / ARN) directamente. La red neuronal convolucional profunda logró resultados de vanguardia en tareas desafiantes como el procesamiento de imágenes, video, audio y habla (Krizhevsky et al., 2012; LeCun et al., 2015; Schmidhuber, 2015; Szegedy et al. , 2015). Además, se aplicó con éxito en problemas biológicos como DeepBind (Alipanahi et al., 2015), DeepCpG (Angermueller et al., 2017), selección de puntos de ramificación (Nazari et al., 2018), predicción de sitios de splicing alternativos (Oubounyt et al., 2018), predicción de sitios de 2 «-Ometilación (Tahir et al., 2018), cuantificación de la secuencia de ADN (Quang y Xie, 2016), localización subcelular de proteínas humanas (Wei et al., 2018), etc. CNN recientemente ganó una atención significativa en la tarea de reconocimiento de promotores. Muy recientemente, Umarov y Solovyev (2017) introdujeron CNNprom para la discriminación de secuencias promotoras cortas, esta arquitectura basada en CNN logró altos resultados en la clasificación de secuencias promotoras y no promotoras. Posteriormente, este modelo fue mejorado por Qian et al. (2018) donde los autores utilizaron un clasificador de máquina de vectores de soporte (SVM) para inspeccionar los elementos de la secuencia promotora más importantes.A continuación, los elementos más influyentes se mantuvieron sin comprimir mientras se comprimían los menos importantes. Este proceso resultó en un mejor desempeño. Recientemente, Umarov et al. Propusieron un modelo de identificación de promotores largos. (2019) en el que los autores se centraron en la identificación de la posición de TSS.
En todos los trabajos mencionados anteriormente, el conjunto negativo se extrajo de regiones no promotoras del genoma. Sabiendo que las secuencias promotoras son ricas exclusivamente en elementos funcionales específicos como TATA-box que se encuentra en –30 ~ –25 bp, GC-Box que se ubica en –110 ~ –80 bp, CAAT-Box que se ubica en – 80 ~ –70 pb, etc. Esto da como resultado una alta precisión de clasificación debido a la gran disparidad entre las muestras positivas y negativas en términos de estructura de secuencia. Además, la tarea de clasificación se vuelve fácil de lograr, por ejemplo, los modelos de CNN solo se basarán en la presencia o ausencia de algunos motivos en sus posiciones específicas para tomar la decisión sobre el tipo de secuencia. Por tanto, estos modelos tienen una precisión / sensibilidad muy baja (alto falso positivo) cuando se prueban en secuencias genómicas que tienen motivos promotores pero no son secuencias promotoras. Es bien sabido que hay más motivos TATAAA en el genoma que los que pertenecen a las regiones promotoras. Por ejemplo, solo la secuencia de ADN del cromosoma 1 humano, ftp://ftp.ensembl.org/pub/release-57/fasta/homo_sapiens/dna/, contiene 151 656 motivos TATAAA. Es más que el número máximo aproximado de genes en el genoma humano total. Como ilustración de este problema, notamos que cuando se prueban estos modelos en secuencias no promotoras que tienen caja TATA, clasifican erróneamente la mayoría de estas secuencias. Por lo tanto, para generar un clasificador robusto, el conjunto negativo debe seleccionarse con cuidado, ya que determina las características que utilizará el clasificador para discriminar las clases. La importancia de esta idea ha sido demostrada en trabajos previos como (Wei et al., 2014). En este trabajo, abordamos principalmente este tema y proponemos un enfoque que integra algunos de los motivos funcionales de clase positiva en la clase negativa para reducir la dependencia del modelo de estos motivos.Utilizamos un modelo CNN combinado con LSTM para analizar las características de la secuencia de promotores eucariotas TATA y no TATA humanos y de ratón y construir modelos computacionales que puedan discriminar con precisión secuencias promotoras cortas de las no promotoras.
Materiales y métodos
2.1. Conjunto de datos
Los conjuntos de datos, que se utilizan para entrenar y probar el predictor de promotor propuesto, se recopilan de humanos y ratones. Contienen dos clases distintivas de los promotores, a saber, los promotores TATA (es decir, las secuencias que contienen la caja TATA) y los promotores no TATA. Estos conjuntos de datos se crearon a partir de la base de datos de promotores eucariotas (EPDnew) (Dreos et al., 2012). El EPDnew es una nueva sección en el conocido conjunto de datos de EPD (Périer et al., 2000) que se anota una colección no redundante de promotores POL II eucariotas donde el sitio de inicio de la transcripción se ha determinado experimentalmente. Proporciona promotores de alta calidad en comparación con la colección de promotores ENSEMBL (Dreos et al., 2012) y es de acceso público en https://epd.epfl.ch//index.php. Descargamos secuencias genómicas del promotor TATA y no TATA para cada organismo de EPDnew. Esta operación dio como resultado la obtención de cuatro conjuntos de datos de promotores, a saber: Human-TATA, Human-non-TATA, Mouse-TATA y Mouse-non-TATA. Para cada uno de estos conjuntos de datos, se construye un conjunto negativo (secuencias no promotoras) con el mismo tamaño que el positivo basándose en el enfoque propuesto como se describe en la siguiente sección. Los detalles sobre el número de secuencias promotoras para cada organismo se dan en la Tabla 1. Todas las secuencias tienen una longitud de 300 pb y se extrajeron de -249 ~ + 50 pb (+1 se refiere a la posición TSS). Como control de calidad, utilizamos una validación cruzada de 5 veces para evaluar el modelo propuesto. En este caso, se utilizan 3 pliegues para entrenamiento, 1 pliegue se usa para validación y el pliegue restante se usa para pruebas. Por lo tanto, el modelo propuesto se entrena 5 veces y se calcula el rendimiento general del 5 veces.

Tabla 1. Estadísticas de los cuatro conjuntos de datos utilizados en este estudio.
2.2. Construcción de conjuntos de datos negativos
Para entrenar un modelo que pueda realizar con precisión la clasificación de secuencias promotoras y no promotoras, debemos elegir cuidadosamente el conjunto negativo (secuencias no promotoras). Este punto es crucial para hacer un modelo capaz de generalizar bien y, por lo tanto, capaz de mantener su precisión cuando se evalúa en conjuntos de datos más desafiantes. Trabajos anteriores, como (Qian et al., 2018), construyeron un conjunto negativo mediante la selección aleatoria de fragmentos de regiones no promotoras del genoma. Obviamente, este enfoque no es completamente razonable porque si no hay una intersección entre conjuntos positivos y negativos. Por lo tanto, el modelo encontrará fácilmente características básicas para separar las dos clases. Por ejemplo, el motivo TATA se puede encontrar en todas las secuencias positivas en una posición específica (normalmente 28 pb aguas arriba del TSS, entre –30 y –25 pb en nuestro conjunto de datos). Por lo tanto, crear un conjunto negativo aleatoriamente que no contenga este motivo producirá un alto rendimiento en este conjunto de datos. Sin embargo, el modelo no clasifica las secuencias negativas que tienen el motivo TATA como promotores. En resumen, el principal defecto de este enfoque es que al entrenar un modelo de aprendizaje profundo, solo aprende a discriminar las clases positivas y negativas en función de la presencia o ausencia de algunas características simples en posiciones específicas, lo que hace que estos modelos sean impracticables. En este trabajo, nuestro objetivo es resolver este problema estableciendo un método alternativo para derivar el conjunto negativo del positivo.
Nuestro método se basa en el hecho de que siempre que las características son comunes entre el negativo y el positivo. clase positiva, el modelo tiende, al tomar la decisión, a ignorar o reducir su dependencia de estas características (es decir, asignar pesos bajos a estas características). En cambio, el modelo se ve obligado a buscar características más profundas y menos obvias. Los modelos de aprendizaje profundo generalmente sufren de una convergencia lenta durante el entrenamiento con este tipo de datos. Sin embargo, este método mejora la robustez del modelo y asegura la generalización. Reconstruimos el conjunto negativo de la siguiente manera. Cada secuencia positiva genera una secuencia negativa. La secuencia positiva se divide en 20 subsecuencias. Luego, se seleccionan 12 subsecuencias al azar y se sustituyen al azar. Se conservan las 8 subsecuencias restantes. Este proceso se ilustra en la Figura 1. La aplicación de este proceso al conjunto positivo da como resultado nuevas secuencias no promotoras con partes conservadas de secuencias promotoras (las subsecuencias sin cambios, 8 subsecuencias de 20). Estos parámetros permiten generar un conjunto negativo que tiene 32 y 40% de sus secuencias conteniendo porciones conservadas de secuencias promotoras. Se encuentra que esta relación es óptima para tener un predictor de promotor robusto como se explica en la sección 3.2.Debido a que las partes conservadas ocupan las mismas posiciones en las secuencias negativas, los motivos obvios como TATA-box y TSS ahora son comunes entre los dos conjuntos con una proporción de 32 ~ 40%. Los logotipos de secuencia de los conjuntos positivo y negativo para los datos del promotor TATA tanto humano como de ratón se muestran en las Figuras 2, 3, respectivamente. Puede verse que los conjuntos positivo y negativo comparten los mismos motivos básicos en las mismas posiciones, como el motivo TATA en la posición -30 y -25 pb y el TSS en la posición +1 pb. Por lo tanto, el entrenamiento es más desafiante pero el modelo resultante se generaliza bien.
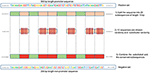
Figura 1. Ilustración del método de construcción de conjuntos negativos. El verde representa las subsecuencias conservadas al azar mientras que el rojo representa las sustituidas y elegidas al azar.

Figura 2. El logotipo de la secuencia en el promotor TATA humano para el conjunto positivo (A) y el conjunto negativo (B). Los gráficos muestran la conservación de los motivos funcionales entre los dos conjuntos.

Figura 3. El logotipo de secuencia en el promotor TATA de ratón para el conjunto positivo (A) y el conjunto negativo (B). Los gráficos muestran la conservación de los motivos funcionales entre los dos conjuntos.
2.3. Los modelos propuestos
Proponemos un modelo de aprendizaje profundo que combina capas de convolución con capas recurrentes como se muestra en la Figura 4. Acepta una sola secuencia genómica sin procesar, S = {N1, N2,…, Nl} donde N ∈ {A, C, G, T} y l es la longitud de la secuencia de entrada, como entrada y salida, una puntuación con valor real. La entrada está codificada en caliente y representada como un vector unidimensional con cuatro canales. La longitud del vector l = 300 y los cuatro canales son A, C, G y T y se representan como (1 0 0 0), (0 1 0 0), (0 0 1 0), (0 0 0 1 ), respectivamente. Para seleccionar el modelo de mejor rendimiento, hemos utilizado el método de búsqueda de cuadrícula para elegir los mejores hiperparámetros. Hemos probado diferentes arquitecturas como CNN solo, LSTM solo, BiLSTM solo, CNN combinado con LSTM. Los hiperparámetros ajustados son el número de capas de convolución, el tamaño del kernel, el número de filtros en cada capa, el tamaño de la capa de agrupación máxima, la probabilidad de abandono y las unidades de la capa Bi-LSTM.
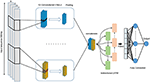
Figura 4. La arquitectura del modelo propuesto de DeePromoter.
El modelo propuesto comienza con múltiples capas de convolución que están alineadas en paralelo y ayudan a aprender los motivos importantes de las secuencias de entrada con diferentes tamaños de ventana. Usamos tres capas de convolución para promotores no TATA con tamaños de ventana de 27, 14 y 7, y dos capas de convolución para promotores TATA con tamaños de ventana de 27, 14. Todas las capas de convolución van seguidas de la función de activación ReLU (Glorot et al. , 2011), una capa de agrupación máxima con un tamaño de ventana de 6 y una capa de abandono con una probabilidad de 0,5. Luego, las salidas de estas capas se concatenan juntas y se introducen en una capa bidireccional de memoria a largo plazo a corto plazo (BiLSTM) (Schuster y Paliwal, 1997) con 32 nodos para capturar las dependencias entre los motivos aprendidos de las capas de convolución. Las funciones aprendidas después de BiLSTM se aplanan y se siguen de abandono con una probabilidad de 0,5. Luego agregamos dos capas completamente conectadas para la clasificación. La primera tiene 128 nodos y le sigue ReLU y abandono con una probabilidad de 0,5, mientras que la segunda capa se utiliza para la predicción con un nodo y función de activación sigmoidea. BiLSTM permite que la información persista y aprenda las dependencias a largo plazo de muestras secuenciales como ADN y ARN. Esto se logra a través de la estructura LSTM que se compone de una celda de memoria y tres puertas llamadas entrada, salida y puertas de olvido. Estas puertas son responsables de regular la información en la celda de memoria. Además, la utilización del módulo LSTM aumenta la profundidad de la red mientras que el número de parámetros requeridos permanece bajo. Tener una red más profunda permite extraer características más complejas y este es el objetivo principal de nuestros modelos, ya que el conjunto negativo contiene muestras duras.
El marco de Keras se utiliza para construir y entrenar los modelos propuestos (Chollet F. et al., 2015). El optimizador Adam (Kingma y Ba, 2014) se utiliza para actualizar los parámetros con una tasa de aprendizaje de 0,001. El tamaño del lote se establece en 32 y el número de épocas se establece en 50. La detención anticipada se aplica en función de la pérdida de validación.
Resultados y discusión
3.1. Medidas de desempeño
En este trabajo, usamos las métricas de evaluación ampliamente adoptadas para evaluar el desempeño de los modelos propuestos.Estas métricas son precisión, recuperación y coeficiente de correlación de Matthew (MCC), y se definen de la siguiente manera:
Donde TP es verdadero positivo y representa secuencias promotoras correctamente identificadas, TN es verdadero negativo y representa secuencias promotoras correctamente rechazadas, FP es falso positivo y representa incorrectamente identificado secuencias promotoras, y FN es falso negativo y representa secuencias promotoras rechazadas incorrectamente.
3.2. Efecto del conjunto negativo
Al analizar los trabajos publicados anteriormente para la identificación de secuencias promotoras, notamos que el rendimiento de esos trabajos depende en gran medida de la forma de preparar el conjunto de datos negativos. Se desempeñaron muy bien en los conjuntos de datos que han preparado, sin embargo, tienen una alta proporción de falsos positivos cuando se evalúan en un conjunto de datos más desafiante que incluye secuencias no apuntadoras que tienen motivos comunes con secuencias promotoras. Por ejemplo, en el caso del conjunto de datos del promotor TATA, las secuencias generadas aleatoriamente no tendrán motivo TATA en la posición -30 y -25 pb, lo que a su vez facilita la tarea de clasificación. En otras palabras, su clasificador dependía de la presencia del motivo TATA para identificar la secuencia promotora y, como resultado, era fácil lograr un alto rendimiento en los conjuntos de datos que habían preparado. Sin embargo, sus modelos fallaron dramáticamente al tratar con secuencias negativas que contenían motivo TATA (ejemplos difíciles). La precisión disminuyó a medida que aumentaba la tasa de falsos positivos. Simplemente, clasificaron estas secuencias como secuencias promotoras positivas. Un análisis similar es válido para los otros motivos promotores. Por lo tanto, el propósito principal de nuestro trabajo no solo es lograr un alto rendimiento en un conjunto de datos específico, sino también mejorar la capacidad del modelo para generalizar bien mediante el entrenamiento en un conjunto de datos desafiante.
Para ilustrar más este punto, entrenamos y probar nuestro modelo en los conjuntos de datos del promotor TATA humano y de ratón con diferentes métodos de preparación de conjuntos negativos. El primer experimento se realiza utilizando secuencias negativas muestreadas al azar de regiones no codificantes del genoma (es decir, similar al enfoque utilizado en los trabajos anteriores). Sorprendentemente, nuestro modelo propuesto logra una precisión de predicción casi perfecta (precisión = 99%, recuperación = 99%, Mcc = 98%) y (precisión = 99%, recuperación = 98%, Mcc = 97%) tanto para humanos como para ratones, respectivamente . Se esperan estos altos resultados, pero la pregunta es si este modelo puede mantener el mismo rendimiento cuando se evalúa en un conjunto de datos que tiene ejemplos concretos. La respuesta, basada en el análisis de los modelos anteriores, es no. El segundo experimento se realiza utilizando nuestro método propuesto para preparar el conjunto de datos como se explica en la sección 2.2. Preparamos los conjuntos negativos que contienen TATA-box conservados con diferentes porcentajes como 12, 20, 32 y 40% y el objetivo es reducir la brecha entre la precisión y la recuperación. Esto asegura que nuestro modelo aprenda características más complejas en lugar de aprender solo la presencia o ausencia de TATA-box. Como se muestra en las Figuras 5A, B, el modelo se estabiliza en la proporción 32 ~ 40% para conjuntos de datos de promotores TATA humanos y de ratón.
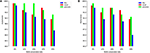
Figura 5. El efecto de diferentes proporciones de conservación del motivo TATA en el conjunto negativo sobre el rendimiento en el caso del conjunto de datos del promotor TATA tanto para humanos (A) como para ratones (B) .
3.3. Resultados y comparación
Durante los últimos años, se han propuesto muchas herramientas de predicción de regiones promotoras (Hutchinson, 1996; Scherf et al., 2000; Reese, 2001; Umarov y Solovyev, 2017). Sin embargo, algunas de estas herramientas no están disponibles públicamente para las pruebas y algunas de ellas requieren más información además de las secuencias genómicas sin procesar. En este estudio, comparamos el desempeño de nuestros modelos propuestos con el trabajo actual de vanguardia, CNNProm, que fue propuesto por Umarov y Solovyev (2017) como se muestra en la Tabla 2. Generalmente, los modelos propuestos, DeePromoter, supere claramente a CNNProm en todos los conjuntos de datos con todas las métricas de evaluación. Más específicamente, DeePromoter mejora la precisión, la recuperación y el MCC en el caso del conjunto de datos TATA humano en 0,18, 0,04 y 0,26, respectivamente. En el caso del conjunto de datos humanos no TATA, DeePromoter mejora la precisión en 0.39, la recuperación en 0.12 y MCC en 0.66. De manera similar, DeePromoter mejora la precisión y MCC en el caso del conjunto de datos TATA de ratón en 0,24 y 0,31, respectivamente. En el caso del conjunto de datos de ratón que no es TATA, DeePromoter mejora la precisión en 0,37, la recuperación en 0,04 y el MCC en 0,65. Estos resultados confirman que CNNProm no rechaza secuencias negativas con el promotor TATA, por lo tanto, tiene un alto número de falsos positivos. Por otro lado, nuestros modelos pueden tratar estos casos con más éxito y la tasa de falsos positivos es menor en comparación con CNNProm.

Tabla 2. Comparación del DeePromoter con el estado de -el método del arte.
Para análisis adicionales, estudiamos el efecto de la alternancia de nucleótidos en cada posición en la puntuación de salida. Nos centramos en la región –40 y 10 pb, ya que alberga la parte más importante de la secuencia promotora. Para cada secuencia promotora en el conjunto de prueba, realizamos un escaneo de mutación computacional para evaluar el efecto de mutar cada base de la subsecuencia de entrada (150 sustituciones en la subsecuencia de intervalo -40 ~ 10 pb). Esto se ilustra en las Figuras 6, 7 para conjuntos de datos TATA humanos y de ratón, respectivamente. El color azul representa una caída en la puntuación de salida debido a la mutación, mientras que el color rojo representa el incremento de la puntuación debido a la mutación. Observamos que la alteración de los nucleótidos a C o G en la región –30 y –25 pb reduce significativamente la puntuación de salida. Esta región es la caja TATA, que es un motivo funcional muy importante en la secuencia promotora. Por lo tanto, nuestro modelo es capaz de encontrar con éxito la importancia de esta región. En el resto de posiciones, los nucleótidos C y G son más preferibles que A y T, especialmente en el caso del ratón. Esto puede explicarse por el hecho de que la región promotora tiene más nucleótidos C y G que A y T (Shi y Zhou, 2006).

Figura 6. El mapa de prominencia de la región –40 bp a 10 bp, que incluye la caja TATA, en el caso de secuencias promotoras TATA humanas.

Figura 7. El mapa de prominencia de la región –40 pb a 10 pb, que incluye la caja TATA, en el caso de secuencias promotoras TATA de ratón.
Conclusión
La predicción precisa de las secuencias promotoras es esencial para comprender el mecanismo subyacente del proceso de regulación génica. En este trabajo, desarrollamos DeePromoter -que se basa en una combinación de red neuronal convolucional y LSTM bidireccional- para predecir las secuencias promotoras eucariotas cortas en el caso de humanos y ratones para promotores TATA y no TATA. El componente esencial de este trabajo fue superar el problema de la baja precisión (alta tasa de falsos positivos) observado en las herramientas desarrolladas anteriormente debido a la dependencia de algunas características / motivos obvios en la secuencia al clasificar las secuencias promotoras y no promotoras. En este trabajo, estábamos particularmente interesados en construir un conjunto negativo duro que impulse a los modelos a explorar la secuencia en busca de características profundas y relevantes en lugar de solo distinguir las secuencias promotoras y no promotoras en función de la existencia de algunos motivos funcionales. Los principales beneficios de usar DeePromoter es que reduce significativamente la cantidad de predicciones falsas positivas al tiempo que logra una alta precisión en conjuntos de datos desafiantes. DeePromoter superó al método anterior no solo en el rendimiento sino también en la superación del problema de las altas predicciones de falsos positivos. Se proyecta que este marco podría ser útil en aplicaciones académicas y relacionadas con las drogas.
Contribuciones de los autores
MO y ZL prepararon el conjunto de datos, concibieron el algoritmo y llevaron a cabo el experimento y análisis. MO y HT prepararon el servidor web y redactaron el manuscrito con el apoyo de ZL y KC. Todos los autores discutieron los resultados y contribuyeron al manuscrito final.
Financiamiento
Esta investigación fue apoyada por el Brain Research Program de la National Research Foundation (NRF) financiado por el gobierno coreano ( MSIT) (No. NRF-2017M3C7A1044815).
Declaración de conflicto de intereses
Los autores declaran que la investigación se realizó en ausencia de relaciones comerciales o financieras que pudieran interpretarse como un posible conflicto de intereses.
Bharanikumar, R., Premkumar, KAR y Palaniappan, A. (2018). Promoterpredict: el modelado basado en secuencias de la fuerza del promotor de escherichia coli σ70 produce una dependencia logarítmica entre la fuerza del promotor y la secuencia. PeerJ 6: e5862. doi: 10.7717 / peerj.5862
Resumen de PubMed | CrossRef Texto completo | Google Académico
Glorot, X., Bordes, A. y Bengio, Y. (2011). «Redes neuronales de rectificador disperso profundo», en Actas de la Decimocuarta Conferencia Internacional sobre Inteligencia Artificial y Estadísticas, (Fort Lauderdale, FL:) 315–323.
Google Scholar
Hutchinson, G. (1996). La predicción de regiones promotoras de vertebrados usando análisis de frecuencia diferencial de hexámeros. Bioinformática 12, 391–398.
Resumen de PubMed | Google Scholar
Kingma, DP y Ba, J. (2014). Adam: un método para la optimización estocástica. arXiv preprint arXiv: 1412.6980.
Google Scholar
Knudsen, S. (1999). Promoter 2. 0: para el reconocimiento de secuencias promotoras de polii. Bioinformatics 15, 356-361.
Resumen de PubMed | Google Scholar
Ponger, L. y Mouchiroud, D. (2002). Cpgprod: identificación de islas cpg asociadas con sitios de inicio de transcripción en grandes secuencias genómicas de mamíferos. Bioinformatics 18, 631–633. doi: 10.1093 / bioinformatics / 18.4.631
Resumen de PubMed | CrossRef Texto completo | Google Académico
Quang, D. y Xie, X. (2016). Danq: una red neuronal profunda convolucional y recurrente híbrida para cuantificar la función de secuencias de adn. Ácidos nucleicos Res. 44, e107 – e107. doi: 10.1093 / nar / gkw226
Resumen de PubMed | CrossRef Texto completo | Google Scholar
Umarov, R. K. y Solovyev, V. V. (2017). Reconocimiento de promotores procariotas y eucariotas utilizando redes neuronales convolucionales de aprendizaje profundo. PLoS ONE 12: e0171410. doi: 10.1371 / journal.pone.0171410
Resumen de PubMed | CrossRef Texto completo | Google Académico