Genetiikan rajat
Johdanto
Promoottorit ovat avainelementtejä, jotka kuuluvat genomin koodaamattomiin alueisiin. Ne hallitsevat suurelta osin geenien aktivaatiota tai tukahduttamista. Ne sijaitsevat geenin transkription aloituskohdan (TSS) lähellä ja ylävirtaan. Geenin promoottorin reunustava alue voi sisältää monia tärkeitä lyhyitä (5 ja 15 emäksen pituisia) DNA-elementtejä ja motiiveja, jotka toimivat tunnistuskohteina proteiineille, jotka tuottavat alavirran geenin transkription asianmukainen aloittaminen ja säätely (Juven-Gershon et ai., 2008). Geenitranskription aloittaminen on perustavanlaatuisin vaihe geeniekspression säätelyssä. Promoottoriydin on minimaalinen DNA-sekvenssi, joka muodostaa TSS: n ja riittää aloittamaan transkription suoraan. Ydinpromoottorin pituus vaihtelee tyypillisesti 60 ja 120 emäsparin välillä (bp).
TATA-ruutu on promoottorisekvenssi, joka osoittaa muille molekyyleille, missä transkriptio alkaa. Se nimettiin ”TATA-boxiksi”, koska sen sekvenssille on ominaista toistuvat T- ja A-emäsparit (TATAAA) (Baker et ai., 2003). Suurin osa TATA-laatikkoa koskevista tutkimuksista on tehty ihmisillä, hiivalla, ja Drosophila-genomeista, mutta samanlaisia elementtejä on löydetty muista lajeista, kuten arkeista ja muinaisista eukaryooteista (Smale ja Kadonaga, 2003) .Ihmisen tapauksessa 24%: lla geeneistä on promoottorialueita, jotka sisältävät TATA-boxia (Yang et al., 2007 Eukaryooteissa TATA-box sijaitsee ~ 25 bp ylävirtaan TSS: stä (Xu et ai., 2016). Se pystyy määrittelemään transkriptiosuunnan ja ilmaisee myös luettavan DNA-juosteen. sitoutua useisiin koodaamattomiin alueisiin, mukaan lukien TATA-box, ja rekrytoida RNA-polymeraasiksi kutsuttu entsyymi, joka syntetisoi RNA: ta DNA: sta.
Promoottorien tärkeän roolin vuoksi geenin transkriptiossa promoottorikohtien tarkka ennustaminen tulee vaadittu vaihe geeniekspressiossa, kuvioiden tulkinnassa sekä rakentamisessa ja ymmärtämisessä geenisääntelyverkkojen toimivuutta. Promoottoreiden tunnistamiseksi oli erilaisia biologisia kokeita, kuten mutaatioanalyysi (Matsumine et ai., 1998) ja immunosaostusmääritykset (Kim et ai., 2004; Dahl ja Collas, 2008). Nämä menetelmät olivat kuitenkin sekä kalliita että aikaa vieviä. Viime aikoina seuraavan sukupolven sekvensoinnin (NGS) (Behjati ja Tarpey, 2013) kehittyessä on sekvensoitu enemmän eri organismien geenejä ja niiden geenielementtejä on tutkittu laskennallisesti (Zhang et al., 2011). Toisaalta NGS-tekniikan innovaatio on johtanut dramaattiseen koko genomin sekvensoinnin kustannusten laskuun, joten enemmän sekvensointitietoja on saatavilla. Tietojen saatavuus houkuttelee tutkijoita kehittämään laskennallisia malleja promoottorin ennustustehtävälle. Se on kuitenkin edelleen keskeneräinen tehtävä, eikä ole olemassa tehokasta ohjelmistoa, joka pystyttäisi ennustamaan promoottoreita tarkasti.
Promoottorien ennustajat voidaan luokitella käytetyn lähestymistavan perusteella kolmeen ryhmään, nimittäin signaalipohjaiseen lähestymistapaan, sisältöpohjaiseen lähestymistapaan ja GpG-pohjainen lähestymistapa. Signaalipohjaiset ennustimet keskittyvät RNA-polymeraasin sitoutumiskohtaan liittyviin promoottorielementteihin ja jättävät huomiotta sekvenssin ei-elementtiset osat. Tämän seurauksena ennustustarkkuus oli heikko eikä tyydyttävä. Esimerkkejä signaalipohjaisista ennustajista ovat: PromoterScan (Prestridge, 1995), joka käytti TATA-laatikon uutettuja ominaisuuksia ja painotettua matriisia transkriptiotekijöiden sitoutumiskohdista lineaarisen erottimen avulla promoottorisekvenssien luokittelemiseksi ei-promoottorisiksi; Promoter2.0 (Knudsen, 1999), joka poimi ominaisuudet eri laatikoista, kuten TATA-Box, CAAT-Box ja GC-Box, ja välitti ne keinotekoisiin hermoverkoihin (ANN) luokitusta varten; NNPP2.1 (Reese, 2001), joka käytti initiaattorielementtiä (Inr) ja TATA-Boxia ominaisuuksien poimintaan ja aikaviiveen neuroverkko luokitukseen, ja Down ja Hubbard (2002), jotka käyttivät TATA-Boxia ja käyttivät merkityksellisiä vektorikoneita (RVM) luokittelijana. Sisältöpohjaiset ennustajat luottivat k-mer-taajuuden laskemiseen suorittamalla k-pituisen ikkunan koko jakson läpi. Nämä menetelmät jättävät kuitenkin huomiotta sekvenssien emäsparien paikkatiedon. Esimerkkejä sisältöpohjaisista ennustajista ovat: PromFind (Hutchinson, 1996), joka käytti k-mer-taajuutta heksameeripromoottoriennusteen suorittamiseen; PromoterInspector (Scherf et ai., 2000), joka tunnisti promoottoreita sisältävät alueet polymeraasi II -promoottorien yhteisen genomisen kontekstin perusteella skannaamalla erityisiä ominaisuuksia, jotka on määritelty vaihtelevan pituisiksi motiiveiksi; MCPromoter1.1 (Ohler et ai., 1999), joka käytti yhtä interpoloitua 5. asteen Markov-ketjua (IMC) promoottorisekvenssien ennustamiseksi.Lopuksi GpG-pohjaiset ennustimet käyttivät GpG-saarten sijaintia promoottorialueena tai ihmisen geenien ensimmäinen eksonialue sisältää yleensä GpG-saaria (Ioshikhes ja Zhang, 2000; Davuluri et ai., 2001; Lander et ai., 2001; Ponger ja Mouchiroud, 2002). Kuitenkin vain 60% promoottoreista sisältää GpG-saaria, joten tämän tyyppisten ennustajien ennustustarkkuus ei koskaan ylittänyt 60%.
Viime aikoina promoottorin ennustamiseen on käytetty sekvenssipohjaisia lähestymistapoja. Yang et ai. (2017) hyödynsi erilaisia ominaisuusuuttostrategioita kaikkein olennaisimpien sekvenssitietojen keräämiseksi tehostaja-promoottori-vuorovaikutusten ennustamiseksi. Lin et ai. (2017) ehdotti sekvenssipohjaista ennustinta, nimeltään ”iPro70-PseZNC”, sigma70-promoottorin tunnistamiseksi prokaryootissa. Samoin Bharanikumar et ai. (2018) ehdotti PromoterPredictia ennustamaan Escherichia coli -promoottorien voimakkuus dynaamisen moniregressiomenetelmän perusteella, jossa sekvenssit esitettiin sijaintipainomatriiseina (PWM). Kanhere ja Bansal (2005) käyttivät promoottori- ja ei-promoottorisekvenssien välisiä eroja DNA-sekvenssin stabiilisuudessa niiden erottamiseksi. Xiao et ai. (2018) esitteli kaksikerroksisen ennustajan nimeltä iPSW (2L) -PseKNC promoottorisekvenssien tunnistamiseksi sekä promoottorien voimakkuudeksi ottamalla sekvensseistä hybridiominaisuuksia.
Kaikki edellä mainitut ennustimet vaativat domeeni- tietoa käsityönä ominaisuuksien luomiseksi. Toisaalta syvälliseen oppimiseen perustuvat lähestymistavat mahdollistavat tehokkaampien mallien rakentamisen käyttämällä suoraan raakatietoja (DNA / RNA-sekvenssejä). Syvä konvoluutioinen neuroverkko saavutti huipputason tulokset haastavissa tehtävissä, kuten kuvan, videon, äänen ja puheen käsittelyssä (Krizhevsky et al., 2012; LeCun et al., 2015; Schmidhuber, 2015; Szegedy et ai. , 2015). Lisäksi sitä sovellettiin menestyksekkäästi biologisissa ongelmissa, kuten DeepBind (Alipanahi et al., 2015), DeepCpG (Angermueller et al., 2017), haarakohdan valinta (Nazari et al., 2018), vaihtoehtoisten silmukointikohteiden ennustaminen (Oubounyt et ai., 2018), 2 ”-metylaatiokohtien ennustaminen (Tahir et ai., 2018), DNA-sekvenssin kvantifiointi (Quang ja Xie, 2016), ihmisen proteiinin alisolujen lokalisointi (Wei et ai., 2018) jne. CNN sai äskettäin huomattavaa huomiota promoottorin tunnistustehtävässä.Vasta äskettäin Umarov ja Solovyev (2017) esittivät CNNpromin lyhyiden promoottorisekvenssien erottamiseksi, tämä CNN-pohjainen arkkitehtuuri saavutti korkeita tuloksia promoottorisekvenssien ja ei-promoottorisekvenssien luokittelussa. Qian ym. (2018), jossa kirjoittajat käyttivät tukivektorikone (SVM) -luokittajaa tärkeimpien promoottorisekvenssielementtien tarkastamiseen.Seuraavaksi tärkeimmät elementit pidettiin pakkaamattomina pakattaen vähemmän tärkeät elementit. Tämä prosessi johti parempaan suorituskykyyn. Äskettäin Umarov et ai. (2019), jossa kirjoittajat keskittyivät TSS-sijainnin tunnistamiseen.
Kaikissa yllä mainituissa teoksissa negatiivinen joukko otettiin genomin ei-promoottorialueilta. Tietäen, että promoottorisekvenssit sisältävät runsaasti vain tiettyjä toiminnallisia elementtejä, kuten TATA-box, joka sijaitsee –30 ~ –25 bp, GC-Box, joka sijaitsee –110 ~ –80 bp, CAAT-Box, joka sijaitsee – 80 ~ 70 bp, jne. Tämä johtaa korkeaan luokitustarkkuuteen johtuen positiivisten ja negatiivisten näytteiden valtavasta erosta sekvenssirakenteen suhteen. Lisäksi luokitustehtävä muuttuu vaivattomaksi saavuttaa, esimerkiksi CNN-mallit luottavat vain joidenkin motiivien läsnäoloon tai puuttumiseen niiden erityisissä paikoissa tehdessään päätöksen sekvenssityypistä. Siten näillä malleilla on erittäin alhainen tarkkuus / herkkyys (suuri väärä positiivinen), kun ne testataan genomisekvensseillä, joilla on promoottorimotiiveja, mutta ne eivät ole promoottorisekvenssejä. On tunnettua, että genomissa on enemmän TATAAA-motiiveja kuin promoottorialueille kuuluvat. Esimerkiksi pelkästään ihmisen kromosomin 1 DNA-sekvenssi, ftp://ftp.ensembl.org/pub/release-57/fasta/homo_sapiens/dna/, sisältää 151 656 TATAAA-motiivia. Se on enemmän kuin arvioitu geenien enimmäismäärä ihmisen koko genomissa. Tämän ongelman havainnollistamiseksi huomaamme, että testattaessa näitä malleja ei-promoottorisekvensseillä, joissa on TATA-laatikko, he luokittelevat suurimman osan näistä sekvensseistä. Siksi vankan luokittelijan luomiseksi negatiivinen joukko on valittava huolellisesti, koska se määrittää ominaisuudet, joita luokittelija käyttää luokkien erottamiseksi. Tämän idean merkitys on osoitettu aikaisemmissa teoksissa, kuten (Wei et al., 2014). Tässä työssä käsittelemme pääasiassa tätä kysymystä ja ehdotamme lähestymistapaa, joka integroi joitain positiivisen luokan toiminnallisia motiiveja negatiiviseen luokkaan vähentääkseen mallin riippuvuutta näistä motiiveista.Hyödynnämme CNN: ää yhdessä LSTM-mallin kanssa analysoimaan ihmisen ja hiiren TATA- ja ei-TATA-eukaryoottisten promoottorien sekvenssiominaisuudet ja rakennamme laskennallisia malleja, jotka voivat erottaa tarkasti lyhyet promoottorisekvenssit ei-promoottorisekvensseistä.
Materiaalit ja menetelmät
2.1. Dataset
Aineistot, joita käytetään ehdotetun promoottoriennusteen harjoittamiseen ja testaamiseen, kerätään ihmisiltä ja hiiriltä. Ne sisältävät kaksi erottavaa promoottoriluokkaa, nimittäin TATA-promoottorit (ts. Sekvenssit, jotka sisältävät TATA-boxia) ja muut kuin TATA-promoottorit. Nämä tietojoukot on rakennettu Eukaryotic Promoter Database (EPDnew) -tietokannasta (Dreos et ai., 2012). EPDnew on uusi osa tunnetun EPD-tietojoukon alla (Périer et ai., 2000), johon on liitetty eukaryoottisten POL II-promoottoreiden ei-redundantti kokoelma, jossa transkription aloituskohta on määritetty kokeellisesti. Se tarjoaa korkealaatuisia promoottoreita verrattuna ENSEMBL-promoottorikokoelmaan (Dreos et al., 2012), ja se on julkisesti saatavilla osoitteessa https://epd.epfl.ch//index.php. Ladasimme EPDnew: ltä kunkin organismin TATA- ja ei-TATA-promoottorin genomisekvenssit. Tämä toimenpide johti neljän promoottorin tietojoukon hankkimiseen: Human-TATA, Human-non-TATA, Mouse-TATA ja Mouse-non-TATA. Kullekin näistä aineistoista rakennetaan negatiivisen joukko (ei-promoottorisekvenssit), joilla on sama koko positiivinen, ehdotetun lähestymistavan perusteella, kuten seuraavassa osassa kuvataan. Yksityiskohdat kunkin organismin promoottorisekvenssien lukumäärästä on annettu taulukossa 1. Kaikkien sekvenssien pituus on 300 bp ja ne uutettiin -249 ~ + 50 bp: sta (+1 viittaa TSS-asemaan). Laadunvalvonnana käytimme viisinkertaista ristivalidointia ehdotetun mallin arvioimiseksi. Tällöin harjoittelussa käytetään 3-kertaisia, validointiin käytetään 1-kertaisia ja testeihin jäljellä olevaa kerrosta. Siksi ehdotettu malli koulutetaan viisi kertaa ja lasketaan 5-kertaisen suorituskyky.

Taulukko 1. Tässä tutkimuksessa käytettyjen neljän tietojoukon tilastot.
2.2. Negatiivisen tietojoukon rakentaminen
Jotta voisimme kouluttaa mallia, joka pystyy suorittamaan tarkasti promoottori- ja ei-promoottorisekvenssit, meidän on valittava negatiivinen joukko (ei-promoottorisekvenssit) huolellisesti. Tämä kohta on ratkaisevan tärkeä, kun tehdään malli, joka kykenee yleistymään hyvin ja pystyy siten säilyttämään tarkkuutensa, kun sitä arvioidaan haastavammissa aineistoissa. Aikaisemmat teokset, kuten (Qian et ai., 2018), rakensivat negatiivisen joukon valitsemalla satunnaisesti fragmentit genomin ei-promoottorialueilta. Ilmeisesti tämä lähestymistapa ei ole täysin kohtuullinen, koska jos positiivisten ja negatiivisten joukkojen välillä ei ole leikkauspistettä. Siten malli löytää helposti perusominaisuudet kahden luokan erottamiseksi toisistaan. Esimerkiksi TATA-motiivi löytyy kaikista positiivisista sekvensseistä tietyssä asemassa (yleensä 28 bp ylävirtaan TSS: stä, välillä -30 ja –25 pb tietojoukossa). Siksi negatiivisen joukon luominen satunnaisesti, joka ei sisällä tätä motiivia, tuottaa korkeaa suorituskykyä tässä aineistossa. Malli ei kuitenkaan pysty luokittelemaan negatiivisia sekvenssejä, joilla on TATA-motiivi promoottoreina. Lyhyesti sanottuna tämän lähestymistavan suurin puute on, että syvällistä oppimismallia koulutettaessa se oppii erottamaan vain positiiviset ja negatiiviset luokat perustuen joidenkin yksinkertaisten ominaisuuksien olemassaoloon tai puuttumiseen tietyissä tehtävissä, mikä tekee näistä malleista mahdotonta. Tässä työssä pyrimme ratkaisemaan tämän ongelman luomalla vaihtoehtoisen menetelmän negatiivisen joukon johtamiseksi positiivisesta.
Menetelmämme perustuu siihen, että aina kun piirteet ovat yhteisiä negatiivisen ja negatiivisen välillä positiivinen luokka, malli pyrkii päätöstä tehdessään jättämään huomiotta tai vähentämään riippuvuuttaan näistä ominaisuuksista (eli osoittamaan näille ominaisuuksille pieniä painoja). Sen sijaan malli on pakko etsiä syvempiä ja vähemmän ilmeisiä piirteitä. Syväoppimismallit kärsivät yleensä hitaasta lähentymisestä tämäntyyppisten tietojen harjoittamisen aikana. Tämä menetelmä parantaa kuitenkin mallin kestävyyttä ja varmistaa yleistymisen. Rekonstruoimme negatiivisen joukon seuraavasti. Jokainen positiivinen sekvenssi tuottaa yhden negatiivisen sekvenssin. Positiivinen sekvenssi on jaettu 20 osaan. Sitten 12 sekvenssiä valitaan satunnaisesti ja korvataan satunnaisesti. Loput 8 jaksoa säilytetään. Tätä prosessia on havainnollistettu kuviossa 1. Tämän prosessin soveltaminen positiivisiin joukkoihin uusissa ei-promoottorisekvensseissä, joissa on konservoituneita osia promoottorisekvensseistä (muuttumattomat alaryhmät, 8 sekvenssiä 20: sta). Nämä parametrit mahdollistavat negatiivisen joukon muodostamisen, jonka sekvensseistä 32 ja 40% sisältävät promoottorisekvenssien konservoituneet osat. Tämän suhteen todetaan olevan optimaalinen vankan promoottorin ennustajan saamiseksi, kuten kappaleessa 3.2 selitetään.Koska konservoidut osat ovat samoissa paikoissa negatiivisissa sekvensseissä, ilmeiset motiivit, kuten TATA-box ja TSS, ovat nyt yhteisiä näiden kahden sarjan välillä suhteella 32 – 40%. Sekä ihmisen että hiiren TATA-promoottoridatan positiivisten ja negatiivisten sarjojen sekvenssilogot on esitetty kuvissa 2, 3, vastaavasti. Voidaan nähdä, että positiivisilla ja negatiivisilla joukkoilla on samat peruskuviot samoissa paikoissa, kuten TATA-motiivi asemissa -30 ja –25 bp ja TSS motiivissa +1 bp. Siksi koulutus on haastavampi, mutta tuloksena oleva malli yleistyy hyvin.
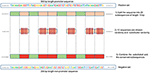
Kuva 1. Kuva negatiivisen joukon muodostusmenetelmästä. Vihreä edustaa satunnaisesti konservoituneita osajoukkoja, kun taas punainen edustaa satunnaisesti valittuja ja korvattuja.

Kuva 2. Sekvenssilogo ihmisen TATA-promoottorissa sekä positiiviselle joukolle (A) että negatiiviselle joukolle (B). Tontit esittävät funktionaalisten motiivien säilyttämistä kahden joukon välillä.

Kuva 3. Sekvenssin logo hiiren TATA-promoottorissa sekä positiiviselle joukolle (A) että negatiiviselle joukolle (B). Käyrät esittävät kahden joukon toiminnallisten motiivien säilyttämistä.
2.3. Ehdotetut mallit
Ehdotamme syvällistä oppimismallia, joka yhdistää konvoluutiokerrokset toistuvien kerrosten kanssa, kuten kuvassa 4 on esitetty. Se hyväksyy yhden raakan genomisekvenssin, S = {N1, N2,…, Nl} missä N ∈ {A, C, G, T} ja l ovat tulosekvenssin pituus, koska tulo ja lähtö antavat reaaliarvotetun pistemäärän. Tulo on yksikoodattu ja esitetty yksiulotteisena vektorina, jossa on neljä kanavaa. Vektorin pituus l = 300 ja neljä kanavaa ovat A, C, G ja T, ja ne esitetään muodossa (1 0 0 0), (0 1 0 0), (0 0 1 0), (0 0 0 1 ). Parhaan suorituskyvyn mallin valitsemiseksi olemme käyttäneet ruudukkohakutapaa parhaiden hyperparametrien valitsemiseksi. Olemme kokeilleet erilaisia arkkitehtuureja, kuten yksinomaan CNN, yksin LSTM, yksin BiLSTM, CNN yhdistettynä LSTM: ään. Viritetyt hyperparametrit ovat konvoluutiokerrosten lukumäärä, ytimen koko, suodattimien lukumäärä kussakin kerroksessa, enimmäispoolikerroksen koko, keskeytymisen todennäköisyys ja Bi-LSTM-kerroksen yksiköt.
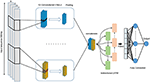
Kuva 4. Ehdotetun DeePromoter-mallin arkkitehtuuri.
Ehdotettu malli alkaa useilla konvoluutiokerroksilla, jotka on kohdistettu rinnakkain ja jotka auttavat oppimaan eri ikkunakokoisten tulosekvenssien tärkeät motiivit. Käytämme kolmea konvoluutiokerrosta ei-TATA-promoottorille, joiden ikkunakoot ovat 27, 14 ja 7, ja kahta konvoluutiokerrosta TATA-promoottoreille, joiden ikkunakoot ovat 27, 14. Kaikkia konvoluutiokerroksia seuraa ReLU-aktivaatiofunktio (Glorot et ai. , 2011), suurin poolikerros, jonka ikkunan koko on 6, ja pudotuskerros todennäköisyydellä 0,5. Sitten näiden kerrosten ulostulot ketjutetaan yhteen ja syötetään kaksisuuntaiseen pitkäaikaisen muistin (BiLSTM) (Schuster ja Paliwal, 1997) kerrokseen, jossa on 32 solmua, jotta vangitaan konvoluutiokerrosten oppimien motiivien väliset riippuvuudet. BiLSTM: n jälkeen opitut piirteet litistetään ja seuraa keskeyttämistä todennäköisyydellä 0,5. Sitten lisätään kaksi täysin yhdistettyä kerrosta luokitusta varten. Ensimmäisessä on 128 solmua ja sitä seuraa ReLU ja keskeyttäminen todennäköisyydellä 0,5, kun taas toista kerrosta käytetään ennustamiseen yhdellä solmulla ja sigmoidilla aktivointitoiminnolla. BiLSTM antaa informaation jatkua ja oppia peräkkäisten näytteiden, kuten DNA: n ja RNA: n, pitkäaikaiset riippuvuudet. Tämä saavutetaan LSTM-rakenteella, joka koostuu muistisolusta ja kolmesta portista, joita kutsutaan tulo-, lähtö- ja unohdusporteiksi. Nämä portit ovat vastuussa muistisolun tiedon säätämisestä. Lisäksi LSTM-moduulin käyttö lisää verkon syvyyttä samalla kun vaadittujen parametrien määrä on edelleen pieni. Syvemmän verkon ansiosta monimutkaisemmat ominaisuudet voidaan poimia, ja tämä on malliemme päätavoite, koska negatiivinen joukko sisältää kovia näytteitä.
Keras-kehystä käytetään ehdotettujen mallien rakentamiseen ja kouluttamiseen (Chollet F. et al., 2015). Adam-optimoijaa (Kingma ja Ba, 2014) käytetään parametrien päivittämiseen oppimisnopeudella 0,001. Erän kooksi on asetettu 32 ja aikakausien määräksi 50. Varhaista pysäytystä käytetään validointihäviön perusteella.
Tulokset ja keskustelu
3.1. Suorituskykymittarit
Tässä työssä käytämme laajalti käytettyjä arviointimittareita ehdotettujen mallien suorituskyvyn arviointiin.Nämä mittarit ovat tarkkuus, palautus ja Matthew-korrelaatiokerroin (MCC), ja ne määritellään seuraavasti:
Jos TP on tosi positiivinen ja edustaa oikein tunnistettuja promoottorisekvenssejä, TN on tosi negatiivinen ja edustaa oikein hylättyjä promoottorisekvenssejä, FP on väärä positiivinen ja edustaa väärin tunnistettuja promoottorisekvenssit ja FN on väärä negatiivinen ja edustaa väärin hylättyjä promoottorisekvenssejä.
3.2. Negatiivisen joukon vaikutus
Kun analysoimme aiemmin julkaistuja teoksia promoottorisekvenssien tunnistamiseksi, huomasimme, että näiden teosten suorituskyky riippuu suuresti tavasta valmistaa negatiivinen aineisto. He suorittivat erittäin hyvin valmistamillaan aineistoilla, mutta heillä on korkea väärä positiivinen suhde, kun ne arvioidaan haastavammalle aineistolle, joka sisältää ei-kehotinsekvenssejä, joilla on yhteisiä motiiveja promoottorisekvenssien kanssa. Esimerkiksi TATA-promoottori-tietojoukon satunnaisilla generoiduilla sekvensseillä ei ole TATA-motiivia asemissa -30 ja –25 bp, mikä puolestaan tekee luokittelusta helpompaa. Toisin sanoen niiden luokittelija riippui TATA-motiivista läsnä ollessa promoottorisekvenssin tunnistamiseksi, ja sen seurauksena oli helppo saavuttaa korkea suorituskyky heidän valmistamillaan aineistoilla. Heidän mallinsa epäonnistuivat kuitenkin dramaattisesti, kun käsiteltiin negatiivisia sekvenssejä, jotka sisälsivät TATA-aiheita (kovat esimerkit). Tarkkuus laski väärän positiivisen määrän kasvaessa. Yksinkertaisesti he luokittelivat nämä sekvenssit positiivisiksi promoottorisekvensseiksi. Samanlainen analyysi pätee muihin promoottorimotiiveihin. Siksi työmme päätarkoitus on paitsi saavuttaa korkea suorituskyky tietyllä tietojoukolla, myös parantaa mallin kykyä yleistää hyvin harjoittamalla haastavaa tietojoukkoa.
Tämän kohdan havainnollistamiseksi koulutamme ja testaa malliamme ihmisen ja hiiren TATA-promoottori-aineistoissa erilaisilla menetelmillä negatiivisten sarjojen valmistamiseksi. Ensimmäinen koe suoritetaan käyttämällä satunnaisesti otettuja negatiivisia sekvenssejä genomin koodaamattomilta alueilta (ts. Samanlainen kuin edellisissä teoksissa käytetty lähestymistapa). Huomattavasti ehdotetulla mallilla saavutetaan lähes täydellinen ennustustarkkuus (tarkkuus = 99%, palautus = 99%, Mcc = 98%) ja (tarkkuus = 99%, palautus = 98%, Mcc = 97%) sekä ihmisellä että hiirellä, vastaavasti . Näitä korkeita tuloksia odotetaan, mutta kysymys on, pystyykö tämä malli säilyttämään saman suorituskyvyn arvioituna tietojoukossa, jolla on kovia esimerkkejä. Aikaisempien mallien analysointiin perustuva vastaus on ei. Toinen koe suoritetaan käyttämällä ehdotettua menetelmää aineiston valmistamiseksi, kuten kohdassa 2.2 selitetään. Valmistelemme negatiiviset sarjat, jotka sisältävät konservoidun TATA-laatikon eri prosenttiosuuksilla, kuten 12, 20, 32 ja 40%, ja tavoitteena on vähentää tarkkuuden ja palautuksen välistä kuilua. Tämä varmistaa, että mallimme oppii monimutkaisempia ominaisuuksia sen sijaan, että opisimme vain TATA-boxin olemassaolon tai puuttumisen. Kuten kuvissa 5A, B on esitetty, malli vakiintuu 32 – 40%: n suhteessa sekä ihmisen että hiiren TATA-promoottori-aineistoihin.
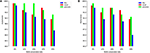
Kuva 5. Negatiivisen joukon TATA-motiivin erilaisten säilyvyyssuhteiden vaikutus suorituskykyyn TATA-promoottoritietojoukossa sekä ihmiselle (A) että hiirelle (B) .
3.3. Tulokset ja vertailu
Viime vuosina on ehdotettu paljon promoottorialueen ennustustyökaluja (Hutchinson, 1996; Scherf et ai., 2000; Reese, 2001; Umarov ja Solovyev, 2017). Jotkut näistä työkaluista eivät kuitenkaan ole julkisesti saatavilla testausta varten, ja jotkut niistä tarvitsevat lisätietoja raakojen genomisekvenssien lisäksi. Tässä tutkimuksessa verrataan ehdotettujen malliemme suorituskykyä nykyiseen huipputekniikkaan, CNNPromiin, jonka Umarov ja Solovyev (2017) ehdottivat taulukon 2 mukaisesti. Yleensä ehdotetut mallit, DeePromoter, ylittää selvästi CNNPromin kaikissa tietojoukoissa kaikilla arviointitiedoilla. Tarkemmin sanottuna DeePromoter parantaa tarkkuutta, palautusta ja MCC: tä ihmisen TATA-tietojoukon kohdalla vastaavasti 0,18, 0,04 ja 0,26. Ihmisen ei-TATA-tietojoukon tapauksessa DeePromoter parantaa tarkkuutta 0,39, palautus 0,12 ja MCC 0,66. Vastaavasti DeePromoter parantaa hiiren TATA-tietojoukon tarkkuutta ja MCC vastaavasti 0,24 ja 0,31. Hiiren, joka ei ole TATA-tietojoukko, DeePromoter parantaa tarkkuutta 0,37, palautus 0,04 ja MCC 0,65. Nämä tulokset vahvistavat, että CNNProm ei hylkää negatiivisia sekvenssejä TATA-promoottorilla, joten sillä on korkea väärä positiivinen. Toisaalta mallimme pystyvät käsittelemään nämä tapaukset paremmin ja väärän positiivisen osuus on pienempi kuin CNNProm.

Taulukko 2. DeePromoterin vertailu tilaan -taide-menetelmä.
Lisäanalyysejä varten tutkitaan vuorotellen nukleotidien vaikutusta kussakin paikassa ulostulopisteisiin. Keskitymme alueeseen –40 ja 10 emäsparia, koska siinä on tärkein osa promoottorisekvenssiä. Kullekin testisarjan promoottorisekvenssille suoritamme laskennallisen mutaation skannauksen arvioidaksemme sisääntulosekvenssin jokaisen emäksen mutaation vaikutuksen (150 substituutiota aikavälillä –40 ~ 10 emäsparia). Tätä havainnollistetaan kuvissa 6, 7 ihmisen ja hiiren TATA-tietojoukoille, vastaavasti. Sininen väri edustaa mutaatiosta johtuvaa lähtöpisteen pudotusta, kun taas punainen väri osoittaa mutaatiosta johtuvaa pisteet. Huomaa, että nukleotidien muuttaminen C: ksi tai G: ksi alueella –30 ja –25 emäsparia vähentää tuotospistettä merkittävästi. Tämä alue on TATA-box, joka on erittäin tärkeä toiminnallinen motiivi promoottorisekvenssissä. Siten malli pystyy onnistuneesti löytämään tämän alueen merkityksen. Muissa asemissa C- ja G-nukleotidit ovat edullisempia kuin A ja T, erityisesti hiiren tapauksessa. Tämä voidaan selittää sillä, että promoottorialueella on enemmän C- ja G-nukleotideja kuin A ja T (Shi ja Zhou, 2006).

Kuva 6. Alueen – 40 – 10 bp: n saliteettikartta, joka sisältää TATA-laatikon, jos kyseessä on ihmisen TATA-promoottorisekvenssi.

Kuva 7. Hiiren TATA-promoottorisekvenssien kohdalla alueen suokartta –40–10 bp, joka sisältää TATA-laatikon.
Päätelmä
Promoottorisekvenssien tarkka ennustaminen on välttämätöntä geenisäätelyprosessin taustalla olevan mekanismin ymmärtämiseksi. Tässä työssä kehitimme DeePromoter-joka perustuu konvoluutio-hermoverkon ja kaksisuuntaisen LSTM-yhdistelmään ennustamaan lyhyet eukaryootti-promoottorisekvenssit ihmisen ja hiiren tapauksessa sekä TATA- että ei-TATA-promoottorille. Tämän työn olennainen osa oli voittaa aiemmin kehitetyissä työkaluissa havaittu matalan tarkkuuden (korkea väärä positiivinen osuus) ongelma, joka johtui siitä, että promoottori- ja ei-promoottorisekvenssejä luokiteltaessa luotettiin sekvenssin joihinkin ilmeisiin piirteisiin / motiiveihin. Tässä työssä olimme erityisen kiinnostuneita rakentamaan kovan negatiivisen joukon, joka ajaa malleja kohti syvällisten ja merkityksellisten ominaisuuksien sekvenssin tutkimista sen sijaan, että erotettaisiin vain promoottori- ja ei-promoottorisekvenssit joidenkin toiminnallisten motiivien olemassaolon perusteella. DeePromoterin käytön tärkeimmät edut ovat, että se vähentää merkittävästi väärien positiivisten ennusteiden määrää ja saavuttaa samalla korkean tarkkuuden haastavissa aineistoissa. DeePromoter ylitti edellisen menetelmän paitsi suorituskyvyssä myös ylittämällä korkean väärän positiivisen ennusteen. Tämän kehyksen ennustetaan olevan hyödyllinen huumeisiin liittyvissä sovelluksissa ja korkeakouluissa.
Kirjoittajan panokset
MO ja ZL valmistivat aineiston, suunnittelivat algoritmin ja suorittivat kokeen ja analyysi. MO ja HT valmistivat verkkopalvelimen ja kirjoittivat käsikirjoituksen ZL: n ja KC: n tuella. Kaikki kirjoittajat keskustelivat tuloksista ja osallistuivat lopulliseen käsikirjoitukseen.
Rahoitus
Tätä tutkimusta tuki Korean hallituksen rahoittama Kansallisen tutkimusrahaston (NRF) aivotutkimusohjelma ( MSIT) (nro NRF-2017M3C7A1044815).
Eturistiriita-ilmoitus
Kirjoittajat ilmoittavat, että tutkimus tehtiin ilman kaupallisia tai taloudellisia suhteita, jotka voitaisiin tulkita mahdollinen eturistiriita.
Bharanikumar, R., Premkumar, KAR ja Palaniappan, A. (2018). Promoottoriennuste: sekvenssipohjainen mallintaminen Escherichia coli σ70 -promoottorivahvuudesta tuottaa logaritmisen riippuvuuden promoottorin voimakkuuden ja sekvenssin välillä. PeerJ 6: e5862. doi: 10.7717 / peerj.5862
PubMed Abstract | CrossRef-kokoteksti | Google Scholar
Glorot, X., Bordes, A. ja Bengio, Y. (2011). ”Deep sparse rectifier neuroverkot”, Proceedings of the Fourteenth International Conference on Artificial Intelligence and Statistics, (Fort Lauderdale, FL:) 315–333.
Google Scholar
Hutchinson, G. (1996). Selkärankaisten promoottorialueiden ennustaminen käyttämällä heksameeritaajuusanalyysiä. Bioinformatiikka 12, 391–398.
PubMed Abstract | Google Scholar
Kingma, DP ja Ba, J. (2014). Adam: menetelmä stokastiseen optimointiin. arXiv-esipainos arXiv: 1412.6980.
Google Scholar
Knudsen, S. (1999). Promoter2. 0: poli-promoottorisekvenssien tunnistamiseksi. Bioinformatiikka 15, 356–361.
PubMed-tiivistelmä | Google Scholar
Ponger, L. ja Mouchiroud, D. (2002). Cpgprod: transkriptioaloituspaikkoihin liittyvien cpg-saarten tunnistaminen suurissa genomisissa nisäkässekvensseissä. Bioinformatiikka 18, 631–633. doi: 10.1093 / bioinformatics / 18.4.631
PubMed Abstract | CrossRef-kokoteksti | Google Scholar
Quang, D. ja Xie, X. (2016). Danq: hybridi konvoluutio- ja toistuva syvä hermoverkko dna-sekvenssien toiminnan kvantifioimiseksi. Nukleiinihapot Res. 44, e107 – e107. doi: 10.1093 / nar / gkw226
PubMed Abstract | CrossRef-kokoteksti | Google Scholar
Umarov, R. K. ja Solovyev, V. V. (2017). Prokaryoottisten ja eukaryoottisten promoottorien tunnistus konvoluutioltaan syvällisesti oppivien hermoverkkojen avulla. PLoS ONE 12: e0171410. doi: 10.1371 / journal.pone.0171410
PubMed Abstract | CrossRef-kokoteksti | Google Scholar