Frontières de la génétique
Introduction
Les promoteurs sont les éléments clés qui appartiennent aux régions non codantes du génome. Ils contrôlent largement l’activation ou la répression des gènes. Ils sont situés à proximité et en amont du site de début de transcription du gène (TSS). La région flanquante du promoteur d’un gène peut contenir de nombreux éléments et motifs d’ADN courts cruciaux (5 et 15 bases de long) qui servent de sites de reconnaissance pour les protéines qui fournissent initiation et régulation appropriées de la transcription du gène en aval (Juven-Gershon et al., 2008). L’initiation de la transcription du gène est l’étape la plus fondamentale dans la régulation de l’expression génique. Le noyau du promoteur est un tronçon minimal de séquence d’ADN qui conations TSS et suffisant pour lancer directement la transcription. La longueur du promoteur de noyau se situe généralement entre 60 et 120 paires de bases (pb).
La boîte TATA est une sous-séquence de promoteur qui indique à d’autres molécules où commence la transcription. Elle a été nommée «TATA-box» car sa séquence est caractérisée par la répétition des paires de bases T et A (TATAAA) (Baker et al., 2003). La grande majorité des études sur la TATA-box ont été menées sur l’homme, la levure, et les génomes de la drosophile, cependant, des éléments similaires ont été trouvés dans d’autres espèces telles que les archées et les eucaryotes anciens (Smale et Kadonaga, 2003). Dans le cas humain, 24% des gènes ont des régions promotrices contenant la boîte TATA (Yang et al., 2007) Chez les eucaryotes, la boîte TATA est située à ~ 25 pb en amont du TSS (Xu et al., 2016). Elle est capable de définir la direction de la transcription et indique également le brin d’ADN à lire. Protéines appelées facteurs de transcription se lient à plusieurs régions non codantes, y compris la boîte TATA et recrutent une enzyme appelée ARN polymérase, qui synthétise l’ARN à partir de l’ADN.
En raison du rôle important des promoteurs dans la transcription génique, la prédiction précise des sites promoteurs devient une étape nécessaire dans l’expression des gènes, l’interprétation des modèles, et la construction et la compréhension la fonctionnalité des réseaux de régulation génétique. Il y a eu différentes expériences biologiques pour l’identification des promoteurs telles que l’analyse mutationnelle (Matsumine et al., 1998) et les tests d’immunoprécipitation (Kim et al., 2004; Dahl et Collas, 2008). Cependant, ces méthodes étaient à la fois coûteuses et longues. Récemment, avec le développement du séquençage de nouvelle génération (NGS) (Behjati et Tarpey, 2013), davantage de gènes de différents organismes ont été séquencés et leurs éléments génétiques ont été explorés par ordinateur (Zhang et al., 2011). D’autre part, l’innovation de la technologie NGS a entraîné une chute spectaculaire du coût du séquençage du génome entier, ainsi, plus de données de séquençage sont disponibles. La disponibilité des données incite les chercheurs à développer des modèles de calcul pour la tâche de prédiction du promoteur. Cependant, il s’agit toujours d’une tâche incomplète et il n’y a pas de logiciel efficace capable de prédire avec précision les promoteurs.
Les prédicteurs de promoteur peuvent être classés en trois groupes en fonction de l’approche utilisée, à savoir l’approche basée sur le signal, l’approche basée sur le contenu et l’approche basée sur GpG. Les prédicteurs basés sur le signal se concentrent sur les éléments promoteurs liés au site de liaison de l’ARN polymérase et ignorent les parties non élémentaires de la séquence. En conséquence, la précision des prévisions était faible et non satisfaisante. Des exemples de prédicteurs basés sur le signal comprennent: PromoterScan (Prestridge, 1995) qui a utilisé les caractéristiques extraites de la boîte TATA et une matrice pondérée de sites de liaison de facteur de transcription avec un discriminateur linéaire pour classer les séquences de promoteur de celles non promoteurs; Promoter2.0 (Knudsen, 1999) qui a extrait les caractéristiques de différentes boîtes telles que TATA-Box, CAAT-Box et GC-Box et les a transmises aux réseaux de neurones artificiels (ANN) pour classification; NNPP2.1 (Reese, 2001) qui a utilisé l’élément initiateur (Inr) et TATA-Box pour l’extraction de caractéristiques et un réseau neuronal à retard pour la classification, et Down et Hubbard (2002) qui ont utilisé TATA-Box et ont utilisé des machines vectorielles de pertinence (RVM) comme classificateur. Les prédicteurs basés sur le contenu reposaient sur le comptage de la fréquence de k-mer en exécutant une fenêtre de k-longueur dans la séquence. Cependant, ces méthodes ignorent les informations spatiales des paires de bases dans les séquences. Des exemples de prédicteurs basés sur le contenu comprennent: PromFind (Hutchinson, 1996) qui a utilisé la fréquence k-mer pour effectuer la prédiction du promoteur hexamère; PromoterInspector (Scherf et al., 2000) qui a identifié les régions contenant des promoteurs sur la base d’un contexte génomique commun des promoteurs de polymérase II en recherchant des caractéristiques spécifiques définies comme des motifs de longueur variable; MCPromoter1.1 (Ohler et al., 1999) qui a utilisé une seule chaîne de Markov interpolée (IMC) du 5ème ordre pour prédire les séquences promotrices.Enfin, les prédicteurs basés sur GpG ont utilisé l’emplacement des îles GpG comme région promotrice ou la première région d’exon dans les gènes humains contient généralement des îles GpG (Ioshikhes et Zhang, 2000; Davuluri et al., 2001; Lander et al., 2001; Ponger et Mouchiroud, 2002). Cependant, seulement 60% des promoteurs contiennent des îlots GpG, donc la précision de prédiction de ce type de prédicteurs n’a jamais dépassé 60%.
Récemment, des approches basées sur des séquences ont été utilisées pour la prédiction des promoteurs. Yang et coll. (2017) ont utilisé différentes stratégies d’extraction de caractéristiques pour capturer les informations de séquence les plus pertinentes afin de prédire les interactions amplificateur-promoteur. Lin et coll. (2017) ont proposé un prédicteur basé sur la séquence, nommé «iPro70-PseZNC», pour l’identification du promoteur sigma70 chez le procaryote. De même, Bharanikumar et al. (2018) ont proposé PromoterPredict afin de prédire la force des promoteurs d’Escherichia coli sur la base d’une approche de régression multiple dynamique où les séquences étaient représentées sous forme de matrices de poids de position (PWM). Kanhere et Bansal (2005) ont utilisé les différences de stabilité de la séquence d’ADN entre les séquences promoteur et non promoteur afin de les distinguer. Xiao et coll. (2018) ont introduit un prédicteur à deux couches appelé iPSW (2L) -PseKNC pour l’identification des séquences de promoteur ainsi que la force des promoteurs en extrayant les caractéristiques hybrides des séquences.
Tous les prédicteurs susmentionnés nécessitent un domaine- connaissances afin de fabriquer les fonctionnalités à la main. D’autre part, les approches basées sur l’apprentissage en profondeur permettent de construire des modèles plus efficaces en utilisant directement des données brutes (séquences ADN / ARN). Le réseau de neurones à convolution profonde a obtenu des résultats de pointe dans des tâches difficiles telles que le traitement de l’image, de la vidéo, de l’audio et de la parole (Krizhevsky et al., 2012; LeCun et al., 2015; Schmidhuber, 2015; Szegedy et al. , 2015). De plus, il a été appliqué avec succès dans des problèmes biologiques tels que DeepBind (Alipanahi et al., 2015), DeepCpG (Angermueller et al., 2017), la sélection de points de branchement (Nazari et al., 2018), la prédiction de sites d’épissage alternatifs (Oubounyt et al., 2018), prédiction des sites de 2 « -Ométhylation (Tahir et al., 2018), quantification de la séquence d’ADN (Quang et Xie, 2016), localisation subcellulaire des protéines humaines (Wei et al., 2018), etc. CNN a récemment attiré une attention significative dans la tâche de reconnaissance des promoteurs. Très récemment, Umarov et Solovyev (2017) ont introduit CNNprom pour la discrimination des séquences de promoteurs courtes, cette architecture basée sur CNN a obtenu des résultats élevés dans la classification des séquences de promoteurs et de non-promoteurs. Par la suite, ce modèle a été amélioré par Qian et al. (2018), où les auteurs ont utilisé un classificateur de machine à vecteurs de support (SVM) pour inspecter les éléments les plus importants de la séquence du promoteur. Ce processus a abouti à de meilleures performances. Récemment, un modèle d’identification de promoteur long a été proposé par Umarov et al. (2019) dans lequel les auteurs se sont concentrés sur l’identification de la position du TSS.
Dans tous les travaux mentionnés ci-dessus, l’ensemble négatif a été extrait de régions non promotrices du génome. Sachant que les séquences promotrices sont riches exclusivement en éléments fonctionnels spécifiques tels que TATA-box qui se situe à –30 ~ –25 bp, GC-Box qui est situé à –110 ~ –80 bp, CAAT-Box qui est situé à – 80 ~ –70 pb, etc. Il en résulte une précision de classification élevée en raison de l’énorme disparité entre les échantillons positifs et négatifs en termes de structure de séquence. De plus, la tâche de classification devient facile à réaliser, par exemple, les modèles CNN se fonderont simplement sur la présence ou l’absence de certains motifs à leurs positions spécifiques pour prendre la décision sur le type de séquence. Ainsi, ces modèles ont une précision / sensibilité très faible (faux positif élevé) lorsqu’ils sont testés sur des séquences génomiques qui ont des motifs promoteurs mais ce ne sont pas des séquences promotrices. Il est bien connu qu’il y a plus de motifs TATAAA dans le génome que ceux appartenant aux régions promotrices. Par exemple, seule la séquence d’ADN du chromosome humain 1, ftp://ftp.ensembl.org/pub/release-57/fasta/homo_sapiens/dna/, contient 151 656 motifs TATAAA. C’est plus que le nombre maximal approximatif de gènes dans le génome humain total. Pour illustrer ce problème, nous remarquons que lors du test de ces modèles sur des séquences non promotrices qui ont une boîte TATA, ils classent mal la plupart de ces séquences. Par conséquent, afin de générer un classificateur robuste, l’ensemble négatif doit être sélectionné avec soin car il détermine les caractéristiques qui seront utilisées par le classificateur afin de discriminer les classes. L’importance de cette idée a été démontrée dans des travaux antérieurs tels que (Wei et al., 2014). Dans ce travail, nous abordons principalement cette question et proposons une approche qui intègre certains des motifs fonctionnels de classe positive dans la classe négative pour réduire la dépendance du modèle vis-à-vis de ces motifs.Nous utilisons un CNN combiné avec un modèle LSTM pour analyser les caractéristiques de séquence des promoteurs eucaryotes humains et murins TATA et non-TATA et construire des modèles de calcul qui peuvent distinguer avec précision les séquences de promoteurs courtes de celles qui ne sont pas des promoteurs.
Matériaux et méthodes
2.1. Ensemble de données
Les ensembles de données, qui sont utilisés pour la formation et le test du prédicteur promoteur proposé, sont collectés auprès de l’homme et de la souris. Ils contiennent deux classes distinctes des promoteurs à savoir les promoteurs TATA (c’est-à-dire les séquences qui contiennent la boîte TATA) et les promoteurs non TATA. Ces ensembles de données ont été construits à partir de la base de données des promoteurs eucaryotes (EPDnew) (Dreos et al., 2012). Le EPDnew est une nouvelle section sous le jeu de données EPD bien connu (Périer et al., 2000) qui est annoté une collection non redondante de promoteurs eucaryotes POL II où le site de début de transcription a été déterminé expérimentalement. Il fournit des promoteurs de haute qualité par rapport à la collection de promoteurs ENSEMBL (Dreos et al., 2012) et il est accessible au public à https://epd.epfl.ch//index.php. Nous avons téléchargé des séquences génomiques de promoteurs TATA et non-TATA pour chaque organisme à partir d’EPDnew. Cette opération a abouti à l’obtention de quatre ensembles de données de promoteur à savoir: Human-TATA, Human-non-TATA, Mouse-TATA et Mouse-non-TATA. Pour chacun de ces ensembles de données, un ensemble négatif (séquences non promotrices) avec la même taille que le positif est construit sur la base de l’approche proposée comme décrit dans la section suivante. Les détails sur le nombre de séquences promotrices pour chaque organisme sont donnés dans le tableau 1. Toutes les séquences ont une longueur de 300 pb et ont été extraites de -249 ~ + 50 pb (+1 se réfère à la position TSS). En tant que contrôle de qualité, nous avons utilisé une validation croisée en 5 fois pour évaluer le modèle proposé. Dans ce cas, 3 fois sont utilisés pour la formation, 1 fois pour la validation et le reste est utilisé pour les tests. Ainsi, le modèle proposé est entraîné 5 fois et les performances globales du 5 fois sont calculées.

Tableau 1. Statistiques des quatre ensembles de données utilisés dans cette étude.
2.2. Construction de l’ensemble de données négatif
Afin de former un modèle capable d’effectuer avec précision la classification des séquences promotrices et non promotrices, nous devons choisir soigneusement l’ensemble négatif (séquences non promotrices). Ce point est crucial pour créer un modèle capable de bien généraliser, et donc capable de maintenir sa précision lorsqu’il est évalué sur des ensembles de données plus complexes. Des travaux antérieurs, tels que (Qian et al., 2018), ont construit un ensemble négatif en sélectionnant au hasard des fragments de régions non promotrices du génome. Évidemment, cette approche n’est pas tout à fait raisonnable car s’il n’y a pas d’intersection entre les ensembles positifs et négatifs. Ainsi, le modèle trouvera facilement des fonctionnalités de base pour séparer les deux classes. Par exemple, le motif TATA peut être trouvé dans toutes les séquences positives à une position spécifique (normalement 28 pb en amont du TSS, entre –30 et –25 pb dans notre jeu de données). Par conséquent, la création aléatoire d’un ensemble négatif qui ne contient pas ce motif produira des performances élevées dans cet ensemble de données. Cependant, le modèle échoue à classer les séquences négatives qui ont le motif TATA comme promoteurs. En bref, le défaut majeur de cette approche est que lors de la formation d’un modèle d’apprentissage en profondeur, il n’apprend qu’à discriminer les classes positives et négatives en fonction de la présence ou de l’absence de certaines caractéristiques simples à des positions spécifiques, ce qui rend ces modèles impraticables. Dans ce travail, nous visons à résoudre ce problème en établissant une méthode alternative pour dériver l’ensemble négatif du positif.
Notre méthode est basée sur le fait que chaque fois que les caractéristiques sont communes entre le négatif et le classe positive le modèle a tendance, lors de la prise de décision, à ignorer ou à réduire sa dépendance vis-à-vis de ces caractéristiques (c’est-à-dire à attribuer de faibles poids à ces caractéristiques). Au lieu de cela, le modèle est obligé de rechercher des fonctionnalités plus profondes et moins évidentes. Les modèles d’apprentissage profond souffrent généralement d’une convergence lente lors de la formation sur ce type de données. Cependant, cette méthode améliore la robustesse du modèle et assure la généralisation. Nous reconstruisons l’ensemble négatif comme suit. Chaque séquence positive génère une séquence négative. La séquence positive est divisée en 20 sous-séquences. Ensuite, 12 sous-séquences sont choisies au hasard et substituées au hasard. Les 8 sous-séquences restantes sont conservées. Ce processus est illustré sur la figure 1. L’application de ce processus à l’ensemble positif conduit à de nouvelles séquences non promotrices avec des parties conservées à partir de séquences promotrices (les sous-séquences inchangées, 8 sous-séquences sur 20). Ces paramètres permettent de générer un ensemble négatif qui a 32 et 40% de ses séquences contenant des portions conservées de séquences promotrices. Ce rapport s’avère optimal pour avoir un prédicteur de promoteur robuste comme expliqué dans la section 3.2.Parce que les parties conservées occupent les mêmes positions dans les séquences négatives, les motifs évidents tels que TATA-box et TSS sont maintenant communs entre les deux ensembles avec un rapport de 32 ~ 40%. Les logos de séquence des ensembles positifs et négatifs pour les données de promoteur TATA humain et murin sont représentés sur les figures 2, 3, respectivement. On peut voir que les ensembles positifs et négatifs partagent les mêmes motifs de base aux mêmes positions comme le motif TATA à la position -30 et –25 pb et le TSS à la position +1 pb. Par conséquent, la formation est plus difficile mais le modèle obtenu se généralise bien.
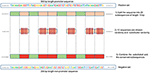
Figure 1. Illustration de la méthode de construction des ensembles négatifs. Le vert représente les sous-séquences conservées au hasard tandis que le rouge représente celles choisies et substituées au hasard.

Figure 2. Le logo de séquence dans le promoteur TATA humain pour à la fois l’ensemble positif (A) et l’ensemble négatif (B). Les graphiques montrent la conservation des motifs fonctionnels entre les deux ensembles.

Figure 3. Le logo de séquence dans le promoteur TATA de la souris pour les ensembles positifs (A) et négatifs (B). Les graphiques montrent la conservation des motifs fonctionnels entre les deux ensembles.
2.3. Les modèles proposés
Nous proposons un modèle d’apprentissage en profondeur qui combine des couches de convolution avec des couches récurrentes comme le montre la figure 4. Il accepte une seule séquence génomique brute, S = {N1, N2,…, Nl} où N ∈ {A, C, G, T} et l est la longueur de la séquence d’entrée, comme entrée et sortie d’un score à valeur réelle. L’entrée est codée à chaud et représentée sous la forme d’un vecteur unidimensionnel avec quatre canaux. La longueur du vecteur l = 300 et les quatre canaux sont A, C, G et T et représentés par (1 0 0 0), (0 1 0 0), (0 0 1 0), (0 0 0 1 ), respectivement. Afin de sélectionner le modèle le plus performant, nous avons utilisé la méthode de recherche par grille pour choisir les meilleurs hyper-paramètres. Nous avons essayé différentes architectures telles que CNN seul, LSTM seul, BiLSTM seul, CNN combiné avec LSTM. Les hyper-paramètres réglés sont le nombre de couches de convolution, la taille du noyau, le nombre de filtres dans chaque couche, la taille de la couche de pooling maximale, la probabilité d’abandon et les unités de la couche Bi-LSTM.
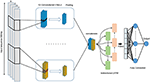
Figure 4. L’architecture du modèle DeePromoter proposé.
Le modèle proposé commence avec plusieurs couches de convolution alignées en parallèle et aident à apprendre les motifs importants des séquences d’entrée avec des tailles de fenêtre différentes. Nous utilisons trois couches de convolution pour un promoteur non TATA avec des tailles de fenêtre de 27, 14 et 7, et deux couches de convolution pour des promoteurs TATA avec des tailles de fenêtre de 27, 14. Toutes les couches de convolution sont suivies par la fonction d’activation ReLU (Glorot et al. , 2011), une couche de pooling max avec une taille de fenêtre de 6 et une couche de suppression d’une probabilité de 0,5. Ensuite, les sorties de ces couches sont concaténées ensemble et introduites dans une couche de mémoire bidirectionnelle à long terme (BiLSTM) (Schuster et Paliwal, 1997) avec 32 nœuds afin de capturer les dépendances entre les motifs appris des couches de convolution. Les caractéristiques apprises après BiLSTM sont aplaties et suivies d’un abandon avec une probabilité de 0,5. Ensuite, nous ajoutons deux couches entièrement connectées pour la classification. Le premier a 128 nœuds et suivi par ReLU et abandon avec une probabilité de 0,5 tandis que la deuxième couche est utilisée pour la prédiction avec un nœud et une fonction d’activation sigmoïde. BiLSTM permet aux informations de persister et d’apprendre les dépendances à long terme d’échantillons séquentiels tels que l’ADN et l’ARN. Ceci est réalisé grâce à la structure LSTM qui est composée d’une cellule mémoire et de trois portes appelées portes d’entrée, de sortie et d’oubli. Ces portes sont chargées de réguler les informations dans la cellule mémoire. De plus, l’utilisation du module LSTM augmente la profondeur du réseau tandis que le nombre de paramètres requis reste faible. Avoir un réseau plus profond permet d’extraire des fonctionnalités plus complexes et c’est l’objectif principal de nos modèles car l’ensemble négatif contient des échantillons durs.
Le framework Keras est utilisé pour construire et entraîner les modèles proposés (Chollet F. et al., 2015). L’optimiseur Adam (Kingma et Ba, 2014) est utilisé pour mettre à jour les paramètres avec un taux d’apprentissage de 0,001. La taille du lot est fixée à 32 et le nombre d’époques est fixé à 50. L’arrêt anticipé est appliqué en fonction de la perte de validation.
Résultats et discussion
3.1. Mesures de performance
Dans ce travail, nous utilisons les métriques d’évaluation largement adoptées pour évaluer les performances des modèles proposés.Ces métriques sont la précision, le rappel et le coefficient de corrélation de Matthew (MCC), et elles sont définies comme suit:
Où TP est vrai positif et représente des séquences promotrices correctement identifiées, TN est vrai négatif et représente des séquences promotrices correctement rejetées, FP est faux positif et représente incorrectement identifié séquences de promoteur, et FN est faux négatif et représente des séquences de promoteur incorrectement rejetées.
3.2. Effet de l’ensemble négatif
Lors de l’analyse des travaux précédemment publiés pour l’identification des séquences promotrices, nous avons remarqué que la performance de ces travaux dépend fortement de la manière de préparer l’ensemble de données négatif. Ils se sont très bien comportés sur les ensembles de données qu’ils ont préparés, cependant, ils ont un taux de faux positifs élevé lorsqu’ils sont évalués sur un ensemble de données plus difficile qui comprend des séquences non prompteurs ayant des motifs communs avec des séquences promotrices. Par exemple, dans le cas de l’ensemble de données du promoteur TATA, les séquences générées aléatoirement n’auront pas de motif TATA à la position -30 et -25 pb, ce qui à son tour facilite la tâche de classification. En d’autres termes, leur classificateur dépendait de la présence du motif TATA pour identifier la séquence du promoteur et, par conséquent, il était facile d’obtenir des performances élevées sur les ensembles de données qu’ils ont préparés. Cependant, leurs modèles ont échoué de façon spectaculaire en traitant des séquences négatives contenant un motif TATA (exemples concrets). La précision diminuait à mesure que le taux de faux positifs augmentait. Simplement, ils ont classé ces séquences comme des séquences promotrices positives. Une analyse similaire est valable pour les autres motifs promoteurs. Par conséquent, l’objectif principal de notre travail n’est pas seulement d’atteindre des performances élevées sur un ensemble de données spécifique, mais également d’améliorer la capacité du modèle à bien généraliser en s’entraînant sur un ensemble de données difficile.
Pour illustrer davantage ce point, nous formons et tester notre modèle sur les ensembles de données du promoteur TATA humain et murin avec différentes méthodes de préparation des ensembles négatifs. La première expérience est réalisée en utilisant des séquences négatives échantillonnées au hasard à partir de régions non codantes du génome (c’est-à-dire, similaire à l’approche utilisée dans les travaux précédents). Remarquablement, notre modèle proposé atteint une précision de prédiction presque parfaite (précision = 99%, rappel = 99%, Mcc = 98%) et (précision = 99%, rappel = 98%, Mcc = 97%) pour l’homme et la souris, respectivement . Ces résultats élevés sont attendus, mais la question est de savoir si ce modèle peut conserver les mêmes performances lorsqu’il est évalué sur un ensemble de données contenant des exemples concrets. La réponse, basée sur l’analyse des modèles précédents, est non. La deuxième expérience est réalisée en utilisant notre méthode proposée pour préparer l’ensemble de données comme expliqué dans la section 2.2. Nous préparons les ensembles négatifs qui contiennent la boîte TATA conservée avec différents pourcentages tels que 12, 20, 32 et 40% et l’objectif est de réduire l’écart entre la précision et le rappel. Cela garantit que notre modèle apprend des fonctionnalités plus complexes plutôt que d’apprendre uniquement la présence ou l’absence de TATA-box. Comme le montrent les figures 5A, B, le modèle se stabilise au rapport 32 ~ 40% pour les ensembles de données de promoteurs TATA humains et souris.
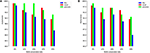
Figure 5. L’effet de différents rapports de conservation du motif TATA dans l’ensemble négatif sur les performances en cas de jeu de données du promoteur TATA pour l’homme (A) et la souris (B) .
3.3. Résultats et comparaison
Au cours des dernières années, de nombreux outils de prédiction des régions promotrices ont été proposés (Hutchinson, 1996; Scherf et al., 2000; Reese, 2001; Umarov et Solovyev, 2017). Cependant, certains de ces outils ne sont pas disponibles publiquement pour les tests et certains d’entre eux nécessitent plus d’informations en plus des séquences génomiques brutes. Dans cette étude, nous comparons les performances de nos modèles proposés avec le travail actuel de pointe, CNNProm, qui a été proposé par Umarov et Solovyev (2017) comme indiqué dans le tableau 2. En général, les modèles proposés, DeePromoter, surpasse clairement CNNProm dans tous les ensembles de données avec toutes les mesures d’évaluation. Plus spécifiquement, DeePromoter améliore la précision, le rappel et le MCC dans le cas de l’ensemble de données TATA humain de 0,18, 0,04 et 0,26, respectivement. Dans le cas d’un jeu de données humain non TATA, DeePromoter améliore la précision de 0,39, le rappel de 0,12 et le MCC de 0,66. De même, DeePromoter améliore la précision et le MCC dans le cas de l’ensemble de données TATA de souris de 0,24 et 0,31, respectivement. Dans le cas d’un ensemble de données non TATA de souris, DeePromoter améliore la précision de 0,37, le rappel de 0,04 et le MCC de 0,65. Ces résultats confirment que CNNProm ne parvient pas à rejeter les séquences négatives avec le promoteur TATA, par conséquent, il a un nombre élevé de faux positifs. D’autre part, nos modèles sont capables de traiter ces cas avec plus de succès et le taux de faux positifs est inférieur à celui de CNNProm.

Tableau 2. Comparaison du DeePromoter avec l’état de -la-méthode de l’art.
Pour des analyses plus poussées, nous étudions l’effet de l’alternance des nucléotides à chaque position sur le score de sortie. Nous nous concentrons sur la région –40 et 10 pb car elle héberge la partie la plus importante de la séquence promotrice. Pour chaque séquence de promoteur dans l’ensemble de test, nous effectuons un balayage de mutation informatique pour évaluer l’effet de la mutation de chaque base de la sous-séquence d’entrée (150 substitutions sur l’intervalle –40 ~ 10 pb sous-séquence). Ceci est illustré sur les figures 6, 7 pour les ensembles de données TATA humains et souris, respectivement. La couleur bleue représente une baisse du score de sortie due à la mutation tandis que la couleur rouge représente l’incrémentation du score due à la mutation. Nous remarquons que la modification des nucléotides en C ou G dans la région –30 et –25 pb réduit considérablement le score de sortie. Cette région est la boîte TATA qui est un motif fonctionnel très important dans la séquence du promoteur. Ainsi, notre modèle parvient à trouver l’importance de cette région. Dans le reste des positions, les nucléotides C et G sont plus préférables que A et T, en particulier dans le cas de la souris. Cela peut s’expliquer par le fait que la région promotrice a plus de nucléotides C et G que A et T (Shi et Zhou, 2006).

Figure 6. La carte de saillance de la région –40 pb à 10 pb, qui inclut la boîte TATA, dans le cas de séquences de promoteurs TATA humains.

Figure 7. La carte de saillance de la région -40 pb à 10 pb, qui inclut la boîte TATA, dans le cas de séquences de promoteurs TATA souris.
Conclusion
Une prédiction précise des séquences du promoteur est essentielle pour comprendre le mécanisme sous-jacent du processus de régulation génique. Dans ce travail, nous avons développé DeePromoter -qui est basé sur une combinaison de réseau de neurones à convolution et de LSTM bidirectionnel- pour prédire les courtes séquences de promoteur eucaryote dans le cas de l’homme et de la souris pour les promoteurs TATA et non-TATA. La composante essentielle de ce travail était de surmonter le problème de la faible précision (taux de faux positifs élevé) remarqué dans les outils précédemment développés en raison de la dépendance à certaines caractéristiques / motifs évidents dans la séquence lors de la classification des séquences promotrices et non promotrices. Dans ce travail, nous nous sommes particulièrement intéressés à la construction d’un ensemble négatif dur qui pousse les modèles vers l’exploration de la séquence pour des caractéristiques profondes et pertinentes au lieu de distinguer uniquement les séquences promotrices et non promotrices basées sur l’existence de certains motifs fonctionnels. Le principal avantage de l’utilisation de DeePromoter est qu’il réduit considérablement le nombre de prédictions faussement positives tout en garantissant une précision élevée sur des ensembles de données difficiles. DeePromoter a surpassé la méthode précédente non seulement en termes de performances, mais aussi en surmontant le problème des prédictions faussement positives élevées. Il est prévu que ce cadre pourrait être utile dans les applications liées à la drogue et dans le milieu universitaire.
Contributions des auteurs
MO et ZL ont préparé le jeu de données, conçu l’algorithme et réalisé l’expérience et Analyse. MO et HT ont préparé le serveur Web et ont rédigé le manuscrit avec le soutien de ZL et KC. Tous les auteurs ont discuté des résultats et ont contribué au manuscrit final.
Financement
Cette recherche a été soutenue par le programme de recherche sur le cerveau de la National Research Foundation (NRF) financé par le gouvernement coréen ( MSIT) (n ° NRF-2017M3C7A1044815).
Déclaration de conflit d’intérêts
Les auteurs déclarent que la recherche a été menée en l’absence de toute relation commerciale ou financière pouvant être interprétée comme un conflit d’intérêts potentiel.
Bharanikumar, R., Premkumar, KAR et Palaniappan, A. (2018). Promoterpredict: la modélisation basée sur la séquence de la force du promoteur d’escherichia coli σ70 produit une dépendance logarithmique entre la force et la séquence du promoteur. PeerJ 6: e5862. doi: 10.7717 / peerj.5862
Résumé PubMed | CrossRef Texte intégral | Google Scholar
Glorot, X., Bordes, A. et Bengio, Y. (2011). «Réseaux de neurones redresseurs clairsemés profonds», dans les actes de la quatorzième conférence internationale sur l’intelligence artificielle et les statistiques, (Fort Lauderdale, FL:) 315–323.
Google Scholar
Hutchinson, G. (1996). La prédiction des régions promotrices des vertébrés à l’aide de l’analyse différentielle des fréquences hexamères. Bioinformatique 12, 391–398.
Résumé PubMed | Google Scholar
Kingma, DP et Ba, J. (2014). Adam: une méthode d’optimisation stochastique. arXiv preprint arXiv: 1412.6980.
Google Scholar
Knudsen, S. (1999). Promoter2. 0: pour la reconnaissance des séquences du promoteur polii. Bioinformatics 15, 356–361.
Résumé PubMed | Google Scholar
Ponger, L. et Mouchiroud, D. (2002). Cpgprod: identification des îlots cpg associés aux sites de début de transcription dans les grandes séquences génomiques de mammifères. Bioinformatics 18, 631–633. doi: 10.1093 / bioinformatics / 18.4.631
Résumé PubMed | CrossRef Texte intégral | Google Scholar
Quang, D. et Xie, X. (2016). Danq: un réseau de neurones profonds hybrides convolutifs et récurrents pour quantifier la fonction des séquences d’ADN. Nucleic Acids Res. 44, e107 à e107. doi: 10.1093 / nar / gkw226
Résumé PubMed | CrossRef Texte intégral | Google Scholar
Umarov, R. K. et Solovyev, V. V. (2017). Reconnaissance de promoteurs procaryotes et eucaryotes à l’aide de réseaux neuronaux convolutifs d’apprentissage profond. PLoS ONE 12: e0171410. doi: 10.1371 / journal.pone.0171410
Résumé PubMed | CrossRef Texte intégral | Google Scholar