Solutions (Français)
Propriétés colligatives
Dépression de la pression de vapeur
Les propriétés physiques peuvent être divisées en deux catégories. Les propriétés étendues (telles que la masse et le volume) dépendent de la taille de l’échantillon. Les propriétés intensives (telles que la densité et la concentration) sont des propriétés caractéristiques de la substance; elles ne dépendent pas de la taille de l’échantillon étudié. Cette section introduit une troisième catégorie qui est un sous-ensemble des propriétés intensives d’un système. Cette troisième catégorie, connue sous le nom de propriétés colligatives, ne peut s’appliquer qu’aux solutions. Par définition, l’une des propriétés d’une solution est une propriété colligative si elle ne dépend que du rapport du nombre de particules de soluté et de solvant dans la solution, et non de l’identité du soluté.
Très peu de propriétés physiques d’une solution sont des propriétés collectives. Comme exemple de cet ensemble limité de propriétés physiques, considérons ce qui arrive à la pression de vapeur du solvant lorsque nous ajoutons un soluté pour former une solution. Nous définirons Po comme la pression de vapeur du liquide pur ![]() le solvant
le solvant ![]() et P comme pression de vapeur du solvant après l’ajout d’un soluté.
et P comme pression de vapeur du solvant après l’ajout d’un soluté.
Po = pression de vapeur du liquide pur, ou du solvant
P = pression de vapeur du solvant en solution
Lorsque la température d’un liquide est inférieure à son ébullition point, nous pouvons supposer que les seules molécules qui peuvent s’échapper du liquide pour former un gaz sont celles qui se trouvent près de la surface du liquide.
Lorsqu’un soluté est ajouté au solvant, certaines des solutémolécules occupent l’espace près de la surface du liquide, comme illustré dans la figure ci-dessous. Lorsqu’un soluté est dissous dans un solvant, le nombre de molécules de solvant près de la surface diminue et la pression de vapeur du solvant diminue.
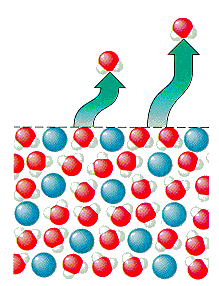
Ceci n’a aucun effet sur la vitesse à laquelle les molécules de solvant en phase gazeuse se condensent pour former un liquide. Mais il diminue le taux auquel les molécules de solvant dans le liquide peuvent s’échapper dans la phase gazeuse. En conséquence, la pression de vapeur du solvant s’échappant d’une solution doit être inférieure à la pression de vapeur du solvant pur.
| P | < | Po | ||
| pression de vapeur du solvant au-dessus d’une solution |
pression de vapeur du solvant pur |
Entre 1887 et 1888, François-Marie Raoult a montré que la pression de vapeur d’une solution est égale à la fraction molaire du solvant multipliée par la pression de vapeur du liquide pur.
| P | = | Csolvent Po | ||||
| pression de vapeur du solvant au-dessus d’une solution |
pression de vapeur du solvant pur |
Cette équation, connue sous le nom de loi de Raoult, est facile à comprendre. Lorsque le solvant est pur et que la fraction molaire du solvant est égale à 1, P est égale à Po. Au fur et à mesure que la fraction molaire du solvant diminue , la pression de vapeur du solvant s’échappant de la solution devient également plus petite.
Supposons, pour le moment, que le solvant est le seul composant de la solution suffisamment volatil pour avoir une pression de vapeur mesurable. Si cela est vrai, la pression de vapeur de la solution sera égale à la pression de vapeur du solvant s’échappant de la solution. La loi de Raoult suggère que la différence entre la pression de vapeur du solvant pur et la solution augmente à mesure que la fraction molaire du solvant diminue.
La variation de la pression de vapeur qui se produit lorsqu’un soluté est ajouté à un solvant est donc une propriété colligative.Si cela dépend de la fraction molaire du soluté, alors il doit dépendre du rapport du nombre de particules de soluté au solvant dans la solution mais pas de l’identité du soluté.
![]()
Élévation du point d’ébullition et dépression du point de congélation
La figure ci-dessous montre les conséquences du fait que les solutés abaissent la pression de vapeur d’un solvant. Le solide Les points de connexion B et C de ce diagramme de phase contiennent les combinaisons de température et de pression auxquelles le solvant pur et sa vapeur sont en équilibre. Chaque point de cette ligne décrit donc la pression de vapeur du purésolvant à cette température. es d’une solution obtenue en dissolvant un soluté dans le solvant.A toute température donnée, la pression de vapeur du solvant s’échappant de la solution est inférieure à la pression de vapeur du solvant pur. La ligne pointillée se trouve donc sous la ligne continue.
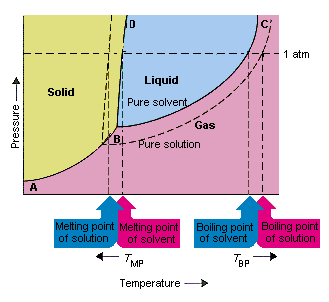
La diminution de la pression de vapeur du solvant qui se produit lorsqu’un soluté est ajouté au solvant provoque une augmentation du point d’ébullition et une diminution du point de fusion de la solution.
D’après cette figure, la solution peut « t bouillir à la même température que le solvant pur. Si la pression de vapeur de le solvant s’échappant de la solution est plus petit que la pression de vapeur du solvant pur à une température donnée, la solution doit être chauffée à une température plus élevée avant qu’elle n’ébullition.L’abaissement de la pression de vapeur du solvant qui se produit lorsqu’il est utilisé pour former une solution donc augmente le point d’ébullition du liquide.
Lorsque les diagrammes de phase ont été introduits, le point triple a été défini comme la seule combinaison de température et de pression à laquelle le gaz, le liquide et le solide peuvent exister en même temps. La figure ci-dessus montre que le point triple de la solution se produit à une température inférieure au point triple du solvant pur.En soi, le changement du point triple n’est pas important. Mais il se traduit par un changement de la température à laquelle la solution gèle ou fond. pourquoi, nous devons faire attention entièrement à la ligne qui sépare les régions solide et liquide dans le diagramme de phase. Cette ligne est presque verticale car le point de fusion d’une substance n’est pas très sensible à la pression.
L’ajout d’un soluté à un solvant ne change pas la façon dont le point de fusion dépend de la pression. La ligne qui sépare les régions solide et liquide de la solution est donc parallèle à la ligne qui remplit la même fonction pour le solvant pur. . Cette ligne doit cependant passer par le point triple de la solution. La diminution du point triple qui se produit quand un soluté est dissous dans un solvant diminue donc le point de fusion de la solution.
La figure ci-dessus montre comment le la variation de la pression de vapeur qui se produit lorsqu’un soluté se dissout dans un solvant entraîne des modifications du point de fusion et du point d’ébullition du solvant, car la variation de la pression de vapeur est une propriété colligative, qui dépend uniquement du nombre relatif de soluté et de particules de solvant , les changements du point d’ébullition et du point de fusion du solvant sont également des propriétés colligatives.
![]()
Calculs ColligativeProperties
La meilleure façon de démons L ‘importance des propriétés colligatives est d’ examiner les conséquences de la loi de Raoult. Raoult a découvert que la pression de vapeur du solvant s’échappant d’une solution est proportionnelle à la fraction molaire du solvant.
P = CsolventPo
Mais la pression de vapeur d’un solvant n’est pas une propriété colligative. Seule la variation de la pression de vapeur qui se produit lorsqu’un soluté est ajouté au solvant peut être incluse parmi les propriétés colligatives d’une solution.
La pression étant une fonction d’état, le changement de pression de vapeur du solvant qui se produit lorsqu’un soluté est ajouté au solvant peut être défini comme la différence entre la pression de vapeur du solvant pur et la pression de vapeur du solvant. s’échapper de la solution.
P = Po – P
La substitution de la loi de Raoult dans cette équation donne le résultat suivant.
P = Po – Csolvent Po = (1 – Csolvent) Po
Cette équation peut être simplifiée en se souvenant de la relation entre la fraction molaire du soluté et la molefraction du solvant.
Csolute + Csolvent = 1
En substituant ceci relation dans l’équation qui définit P donne une autre forme de loi de Raoult.
P = CsolutePo
Cette équation nous rappelle que la variation de la pression de vapeur du solvant qui se produit lorsqu’un soluté est ajouté au solvant est proportionnelle à la fraction molaire du soluté. Au fur et à mesure que le solvant est dissous dans le solvant, la pression de vapeur du solvant diminue et la variation de la pression de vapeur du solvant augmente.
Étant donné que les changements du point d’ébullition du solvant (TBP) qui se produisent lorsqu’un soluté est ajouté à un solvant résultent de changements dans la pression de vapeur du solvant, l’ampleur du changement du point d’ébullition est également proportionnelle. à la molefraction du soluté.
![]() TBP = kbsolute
TBP = kbsolute
Dans les solutions diluées, la fraction molaire du soluté est proportionnelle à la molalité de la solution, comme indiqué dans la figure ci-dessous.

L’équation qui décrit l’amplitude de l’élévation du point d’ébullition qui se produit lorsqu’un soluté est ajouté à un solvant est donc souvent écrite comme suit.
![]() TBP = kbm
TBP = kbm
Ici, ![]() TBP est l’élévation du point d’ébullition – le changement du point d’ébullition qui se produit lorsqu’un soluté se dissout dans le solvant
TBP est l’élévation du point d’ébullition – le changement du point d’ébullition qui se produit lorsqu’un soluté se dissout dans le solvant ![]() et kb est une constante de proportionnalité connue sous le nom de constante d’élévation du point d’ébullition molaire pour le solvant.
et kb est une constante de proportionnalité connue sous le nom de constante d’élévation du point d’ébullition molaire pour le solvant.
Une équation similaire peut être écrite pour décrire ce qui se passe au point de congélation (ou point de fusion) d’un solvant lorsqu’un soluté est ajouté au solvant.
![]() TFP = -kfm
TFP = -kfm
Dans cette équation, ![]() TFP est le point de congélation dépression
TFP est le point de congélation dépression ![]() le changement du point de congélation qui se produit lorsque le soluté se dissout dans le solvant – et kf est la constante de dépression du point de congélation molaire pour le solvant. Un signe négatif est utilisé dans cette équation pour indiquer que le point de congélation du solvant diminue lorsqu’un soluté est ajouté.
le changement du point de congélation qui se produit lorsque le soluté se dissout dans le solvant – et kf est la constante de dépression du point de congélation molaire pour le solvant. Un signe négatif est utilisé dans cette équation pour indiquer que le point de congélation du solvant diminue lorsqu’un soluté est ajouté.
Les valeurs de kf et kbas ainsi que les points de congélation et d’ébullition pour un certain nombre de solvants purs sont donnés dans les tableaux ci-dessous.
Constantes de dépression du point de congélation
Constantes d’élévation du point d’ébullition
Problème pratique 6:
Calculez le poids moléculaire du soufre si 35,5 grammes de soufre se dissolvent dans 100,0 grammes de CS2 pour produire une solution qui a un point d’ébullition de 49,48 ° C.
Cliquez ici pour vérifier votre réponse au problème pratique 6
Cliquez ici pour voir une solution au problème pratique 6
Pratique Problème 7:
Déterminez le poids moléculaire de l’acide acétique si une solution contenant 30,0 grammes d’acide acétique par kilogramme d’eau gèle à -0,93 ° C. Ces résultats sont-ils en accord avec l’hypothèse selon laquelle l’acide acétique répond à la formule CH3CO2H?
Cliquez ici pour vérifier votre réponse au problème pratique 7
Cliquez ici pour voir une solution au problème pratique 7
Que se passerait-il dans le calcul de la pratique Le problème 7 a été répété avec un acide plus fort, tel que l’acide chlorhydrique?
Pratique Problème 8:
Expliquez pourquoi une solution de 0,100 m de HCl dissous dans du benzène a une dépression du point de congélation de 0,512 ° C, tandis qu’une solution de 0,100 m de HCl dans l’eau a une dépression du point de congélation de 0,352 ° C.
Cliquez ici pour vérifier votre réponse au problème pratique 8
En 1884 Jacobus Henricus van « t Hoff a introduit un autre terme dans les expressions de dépression du point de congélation et d’élévation du point d’ébullition pour expliquer les propriétés colligatives des solutions de composés qui se dissocient lorsqu’ils se dissolvent dans l’eau.
![]() TFP = – kf (i) m
TFP = – kf (i) m
En remplaçant la valeur expérimentale de la dépression du point de congélation d’une solution de HCl à 0,100 m dans cette équation, on obtient une valeur pour le terme i de 1,89. Si HCl ne se dissociait pas dans l’eau, i serait 1. S’il se dissocie complètement, je serais 2. La valeur expérimentale de 1,89 suggère qu’au moins 95% des molécules de HCl se dissocient dans cette solution.
Pratique Problème 9:
Expliquez pourquoi 0,60 gramme d’acide acétique se dissout dans 200 grammes de benzène pour former une solution qui abaisse le point de congélation du benzène à 5,40 ° C.
Cliquez ici pour vérifier votre réponse à Problème pratique 9
![]()
Pression osmotique
En 1784, le physicien et ecclésiastique français Jean AntoineNollet découvrit qu’une vessie de porc remplie d’une solution concentrée d’alcool dans l’eau se dilatait lorsqu’elle était immergée dans l’eau. La vessie agissait comme une membrane semi-perméable, qui permettait aux molécules d’eau de pénétrer dans la solution, mais empêchait les molécules d’alcool de se déplacer dans l’autre direction. Le mouvement d’un composant d’une solution à travers une membrane pour diluer la solution est appelé osmose, et la pression qu’elle produit est appelée pression osmotique ().
La pression osmotique peut être démontrée avec l’appareil montré dans la figure ci-dessous. Une membrane semi-perméable est attachée à travers l’extrémité ouverte d’un tube de chardon. Le tube est ensuite partiellement rempli d’une solution de sucre ou d’alcool dans de l’eau et immergé dans un abaisseur d’eau. L’eau s’écoulera dans le tube jusqu’à ce que la pression sur la colonne d’eau due à la force de gravité équilibre la pression osmotique entraînant l’eau à travers la membrane.
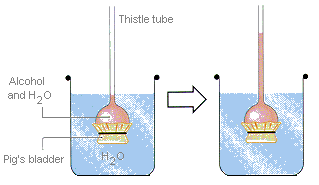
L’eau s’écoule la membrane semi-perméable pour diluer la solution alcoolique jusqu’à ce que la force de gravité tirant vers le bas sur la colonne de cette solution équilibre la pression osmotique poussant l’eau à travers la membrane.
La même année que Raoult a découvert la relation entre la pression de vapeur d’une solution et la pression de vapeur d’un purésolvant, Jacobus Henricus van « t Hoff a constaté que la pression osmotique d’une solution diluée () obéissait à une équation analogue à l’équation des gaz parfaits.
| |
= | nRT | ||
| V |
Cette équation suggère que la pression osmotique est un autre exemple de propriété colligative, car cela la pression dépend du rapport du nombre de particules de soluté au volume de la solution ![]() n / V
n / V ![]() et non de l’identité des solutéparticules. nous rappelle également la magnitude de la pression osmotique. Selon cette équation, un 1,00 M s La solution a une pression osmotique de 22,4 atm à 0 ° C.
et non de l’identité des solutéparticules. nous rappelle également la magnitude de la pression osmotique. Selon cette équation, un 1,00 M s La solution a une pression osmotique de 22,4 atm à 0 ° C.
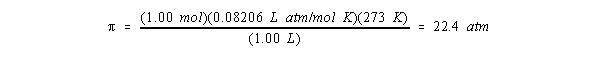
Cela signifie qu’une solution de 1,00 m devrait pouvoir supporter une colonne d’eau de 670 pouces, soit près de 56 pieds de hauteur !
Les biologistes et les biochimistes profitent souvent de la pression osmotique lorsqu’ils isolent les composants d’une cellule. Lorsqu’une cellule est ajoutée à une solution aqueuse qui contient une concentration d’ions beaucoup plus élevée que le liquide à l’intérieur de la cellule, l’eau quitte la cellule en s’écoulant à travers la membrane cellulaire jusqu’à ce que la cellule se rétrécisse tellement que la membrane se brise. Alternativement, lorsqu’une cellule est placée dans une solution qui a une force ionique beaucoup plus petite, l’eau se déverse dans la cellule et la cellule se dilate jusqu’à ce que la membrane cellulaire éclate.
![]()