Grenzen in der Genetik
Einführung
Promotoren sind die Schlüsselelemente, die zu nichtkodierenden Regionen im Genom gehören. Sie steuern weitgehend die Aktivierung oder Repression der Gene. Sie befinden sich in der Nähe und stromaufwärts der Transkriptionsstartstelle (TSS) des Gens. Die Promotor-flankierende Region eines Gens kann viele wichtige kurze DNA-Elemente und Motive (5 und 15 Basen lang) enthalten, die als Erkennungsstellen für die bereitgestellten Proteine dienen ordnungsgemäße Initiierung und Regulation der Transkription des nachgeschalteten Gens (Juven-Gershon et al., 2008). Die Initiierung des Gentranskripts ist der grundlegendste Schritt bei der Regulation der Genexpression. Der Promotor-Kern ist ein minimaler Abschnitt der DNA-Sequenz, der TSS konationiert und ausreicht, um die Transkription direkt zu initiieren. Die Länge des Kernpromotors liegt typischerweise zwischen 60 und 120 Basenpaaren (bp).
Die TATA-Box ist eine Promotor-Subsequenz, die anderen Molekülen anzeigt, wo die Transkription beginnt. Es wurde „TATA-Box“ genannt, da seine Sequenz durch Wiederholung von T- und A-Basenpaaren (TATAAA) gekennzeichnet ist (Baker et al., 2003). Die überwiegende Mehrheit der Studien an der TATA-Box wurde an Menschen, Hefen, und Drosophila-Genome wurden jedoch ähnliche Elemente in anderen Arten wie Archaeen und alten Eukaryoten gefunden (Smale und Kadonaga, 2003). Im menschlichen Fall haben 24% der Gene Promotorregionen, die TATA-Box enthalten (Yang et al., 2007) ) In Eukaryoten befindet sich die TATA-Box bei ~ 25 bp stromaufwärts des TSS (Xu et al., 2016). Sie kann die Transkriptionsrichtung definieren und zeigt auch den zu lesenden DNA-Strang an. Proteine, sogenannte Transkriptionsfaktoren binden an mehrere nicht-kodierende Regionen, einschließlich TATA-Box, und rekrutieren ein Enzym namens RNA-Polymerase, das RNA aus DNA synthetisiert.
Aufgrund der wichtigen Rolle der Promotoren bei der Gentranskription wird eine genaue Vorhersage der Promotorstellen Ein erforderlicher Schritt bei der Genexpression, der Interpretation von Mustern sowie beim Aufbau und Verstehen die Funktionalität genetischer regulatorischer Netzwerke. Es gab verschiedene biologische Experimente zur Identifizierung von Promotoren wie Mutationsanalyse (Matsumine et al., 1998) und Immunpräzipitationstests (Kim et al., 2004; Dahl und Collas, 2008). Diese Methoden waren jedoch sowohl teuer als auch zeitaufwändig. Mit der Entwicklung der Next-Generation-Sequenzierung (NGS) (Behjati und Tarpey, 2013) wurden kürzlich mehr Gene verschiedener Organismen sequenziert und ihre Genelemente rechnerisch untersucht (Zhang et al., 2011). Andererseits hat die Innovation der NGS-Technologie zu einem dramatischen Rückgang der Kosten für die gesamte Genomsequenzierung geführt, sodass mehr Sequenzierungsdaten verfügbar sind. Die Datenverfügbarkeit zieht Forscher an, Rechenmodelle für die Aufgabe der Promotorvorhersage zu entwickeln. Es ist jedoch immer noch eine unvollständige Aufgabe, und es gibt keine effiziente Software, die Promotoren genau vorhersagen kann.
Promotor-Prädiktoren können basierend auf dem verwendeten Ansatz in drei Gruppen eingeteilt werden, nämlich signalbasierten Ansatz, inhaltsbasierten Ansatz und der GpG-basierte Ansatz. Signalbasierte Prädiktoren konzentrieren sich auf Promotorelemente, die mit der RNA-Polymerase-Bindungsstelle zusammenhängen, und ignorieren die Nichtelementteile der Sequenz. Infolgedessen war die Vorhersagegenauigkeit schwach und nicht zufriedenstellend. Beispiele für signalbasierte Prädiktoren umfassen: PromoterScan (Prestridge, 1995), der die extrahierten Merkmale der TATA-Box und eine gewichtete Matrix von Transkriptionsfaktor-Bindungsstellen mit einem linearen Diskriminator verwendete, um Promotorsequenzen von Nicht-Promotorsequenzen zu klassifizieren; Promoter2.0 (Knudsen, 1999), der die Merkmale aus verschiedenen Boxen wie TATA-Box, CAAT-Box und GC-Box extrahierte und zur Klassifizierung an künstliche neuronale Netze (ANN) weitergab; NNPP2.1 (Reese, 2001), das Initiatorelement (Inr) und TATA-Box zur Merkmalsextraktion und ein zeitverzögertes neuronales Netzwerk zur Klassifizierung verwendete, und Down und Hubbard (2002), die TATA-Box verwendeten und Relevanzvektormaschinen verwendeten (RVM) als Klassifikator. Inhaltsbasierte Prädiktoren stützten sich darauf, die Häufigkeit von k-mer zu zählen, indem ein k-Längenfenster über die Sequenz geführt wurde. Diese Methoden ignorieren jedoch die räumlichen Informationen der Basenpaare in den Sequenzen. Beispiele für inhaltsbasierte Prädiktoren umfassen: PromFind (Hutchinson, 1996), das die k-mer-Frequenz verwendete, um die Hexamer-Promotor-Vorhersage durchzuführen; PromoterInspector (Scherf et al., 2000), der die Regionen, die Promotoren enthalten, basierend auf einem gemeinsamen genomischen Kontext von Polymerase II-Promotoren durch Scannen nach spezifischen Merkmalen identifizierte, die als Motive variabler Länge definiert sind; MCPromoter1.1 (Ohler et al., 1999), der eine einzelne interpolierte Markov-Kette (IMC) 5. Ordnung verwendete, um Promotorsequenzen vorherzusagen.Schließlich verwendeten GpG-basierte Prädiktoren die Position von GpG-Inseln als Promotorregion oder die erste Exonregion in den menschlichen Genen, die üblicherweise GpG-Inseln enthalten (Ioshikhes und Zhang, 2000; Davuluri et al., 2001; Lander et al., 2001; Ponger und Mouchiroud, 2002). Allerdings enthalten nur 60% der Promotoren GpG-Inseln, weshalb die Vorhersagegenauigkeit dieser Art von Prädiktoren 60% nie überschritt. In letzter Zeit wurden sequenzbasierte Ansätze zur Promotorvorhersage verwendet. Yang et al. (2017) verwendeten verschiedene Merkmalsextraktionsstrategien, um die relevantesten Sequenzinformationen zu erfassen, um Enhancer-Promotor-Wechselwirkungen vorherzusagen. Lin et al. (2017) schlugen einen sequenzbasierten Prädiktor namens „iPro70-PseZNC“ für die Identifizierung des Sigma70-Promotors im Prokaryoten vor. Ebenso haben Bharanikumar et al. (2018) schlugen PromoterPredict vor, um die Stärke von Escherichia coli-Promotoren basierend auf einem dynamischen multiplen Regressionsansatz vorherzusagen, bei dem die Sequenzen als Positionsgewichtsmatrizen (PWM) dargestellt wurden. Kanhere und Bansal (2005) nutzten die Unterschiede in der DNA-Sequenzstabilität zwischen den Promotor- und Nicht-Promotor-Sequenzen, um sie zu unterscheiden. Xiao et al. (2018) führten einen zweischichtigen Prädiktor namens iPSW (2L) -PseKNC zur Identifizierung von Promotorsequenzen sowie zur Stärke der Promotoren durch Extraktion von Hybridmerkmalen aus den Sequenzen ein.
Alle oben genannten Prädiktoren erfordern Domänen- Wissen, um die Funktionen von Hand herzustellen. Auf der anderen Seite ermöglichen Deep-Learning-basierte Ansätze die Erstellung effizienterer Modelle unter Verwendung von Rohdaten (DNA / RNA-Sequenzen) direkt. Ein tiefes Faltungs-Neuronales Netzwerk erzielte auf dem neuesten Stand der Technik herausfordernde Aufgaben wie die Verarbeitung von Bild, Video, Audio und Sprache (Krizhevsky et al., 2012; LeCun et al., 2015; Schmidhuber, 2015; Szegedy et al. , 2015). Darüber hinaus wurde es erfolgreich bei biologischen Problemen wie DeepBind (Alipanahi et al., 2015), DeepCpG (Angermueller et al., 2017), Auswahl von Verzweigungspunkten (Nazari et al., 2018) und Vorhersage alternativer Spleißstellen (Oubounyt) angewendet et al., 2018), Vorhersage von 2 „-Omethylierungsstellen (Tahir et al., 2018), Quantifizierung der DNA-Sequenz (Quang und Xie, 2016), subzelluläre Lokalisierung von menschlichem Protein (Wei et al., 2018) usw. CNN hat kürzlich erhebliche Aufmerksamkeit bei der Promotorerkennungsaufgabe erlangt. Vor kurzem haben Umarov und Solovyev (2017) CNNprom zur Unterscheidung kurzer Promotorsequenzen eingeführt. Diese CNN-basierte Architektur erzielte hohe Ergebnisse bei der Klassifizierung von Promotor- und Nicht-Promotorsequenzen. Danach wurde dieses Modell verbessert von Qian et al. (2018), wo die Autoren einen SVM-Klassifikator (Support Vector Machine) verwendeten, um die wichtigsten Promotorsequenzelemente zu untersuchen. Als nächstes wurden die einflussreichsten Elemente unkomprimiert gehalten, während die weniger wichtigen komprimiert wurden. Dieser Prozess führte zu einer besseren Leistung. Kürzlich wurde von Umarov et al. Ein Modell zur Identifizierung langer Promotoren vorgeschlagen. (2019), in dem sich die Autoren auf die Identifizierung der TSS-Position konzentrierten.
In allen oben genannten Arbeiten wurde der negative Satz aus Nicht-Promotor-Regionen des Genoms extrahiert. In dem Wissen, dass die Promotorsequenzen ausschließlich an spezifischen funktionellen Elementen reich sind, wie TATA-Box, die sich bei –30 ~ –25 bp befindet, GC-Box, die sich bei –110 ~ –80 bp befindet, CAAT-Box, die sich bei – befindet 80 ~ –70 bp usw. Dies führt zu einer hohen Klassifizierungsgenauigkeit aufgrund der großen Unterschiede zwischen den positiven und negativen Proben hinsichtlich der Sequenzstruktur. Zusätzlich wird die Klassifizierungsaufgabe mühelos zu erreichen, zum Beispiel werden die CNN-Modelle nur auf das Vorhandensein oder Fehlen einiger Motive an ihren spezifischen Positionen angewiesen sein, um die Entscheidung über den Sequenztyp zu treffen. Daher weisen diese Modelle eine sehr geringe Präzision / Empfindlichkeit (hohes falsch positives Ergebnis) auf, wenn sie an Genomsequenzen getestet werden, die Promotormotive aufweisen, jedoch keine Promotorsequenzen sind. Es ist bekannt, dass das Genom mehr TATAAA-Motive enthält als diejenigen, die zu den Promotorregionen gehören. Beispielsweise enthält allein die DNA-Sequenz des menschlichen Chromosoms 1, ftp://ftp.ensembl.org/pub/release-57/fasta/homo_sapiens/dna/, 151 656 TATAAA-Motive. Es ist mehr als die ungefähre maximale Anzahl von Genen im gesamten menschlichen Genom. Zur Veranschaulichung dieses Problems stellen wir fest, dass beim Testen dieser Modelle an Nicht-Promotor-Sequenzen mit TATA-Box die meisten dieser Sequenzen falsch klassifiziert werden. Um einen robusten Klassifizierer zu erzeugen, sollte daher die negative Menge sorgfältig ausgewählt werden, da sie die Merkmale bestimmt, die vom Klassifizierer verwendet werden, um die Klassen zu unterscheiden. Die Bedeutung dieser Idee wurde in früheren Arbeiten wie (Wei et al., 2014) gezeigt. In dieser Arbeit befassen wir uns hauptsächlich mit diesem Problem und schlagen einen Ansatz vor, der einige der Funktionsmotive der positiven Klasse in die negative Klasse integriert, um die Abhängigkeit des Modells von diesen Motiven zu verringern.Wir verwenden ein CNN in Kombination mit einem LSTM-Modell, um die Sequenzmerkmale von eukaryotischen TATA- und Nicht-TATA-Promotoren von Mensch und Maus zu analysieren und Rechenmodelle zu erstellen, mit denen kurze Promotorsequenzen von Nicht-Promotorsequenzen genau unterschieden werden können.
Materialien und Methoden
2.1. Datensatz
Die Datensätze, die zum Trainieren und Testen des vorgeschlagenen Promotor-Prädiktors verwendet werden, werden von Mensch und Maus gesammelt. Sie enthalten zwei unterschiedliche Klassen der Promotoren, nämlich TATA-Promotoren (d. H. Die Sequenzen, die TATA-Box enthalten) und Nicht-TATA-Promotoren. Diese Datensätze wurden aus der Eukaryotic Promoter Database (EPDnew) erstellt (Dreos et al., 2012). Das EPDnew ist ein neuer Abschnitt unter dem bekannten EPD-Datensatz (Périer et al., 2000), der eine nicht redundante Sammlung von eukaryotischen POL II-Promotoren kommentiert, bei denen die Transkriptionsstartstelle experimentell bestimmt wurde. Es bietet im Vergleich zur ENSEMBL-Promotorsammlung (Dreos et al., 2012) qualitativ hochwertige Promotoren und ist unter https://epd.epfl.ch//index.php öffentlich zugänglich. Wir haben genomische Sequenzen von TATA- und Nicht-TATA-Promotoren für jeden Organismus von EPDnew heruntergeladen. Diese Operation führte zum Erhalt von vier Promotor-Datensätzen, nämlich: Human-TATA, Human-Non-TATA, Mouse-TATA und Mouse-Non-TATA. Für jeden dieser Datensätze wird ein negativer Satz (Nicht-Promotor-Sequenzen) mit der gleichen Größe wie der positive auf der Grundlage des vorgeschlagenen Ansatzes konstruiert, wie im folgenden Abschnitt beschrieben. Die Details zur Anzahl der Promotorsequenzen für jeden Organismus sind in Tabelle 1 angegeben. Alle Sequenzen haben eine Länge von 300 bp und wurden aus -249 ~ + 50 bp extrahiert (+1 bezieht sich auf die TSS-Position). Als Qualitätskontrolle verwendeten wir eine 5-fache Kreuzvalidierung, um das vorgeschlagene Modell zu bewerten. In diesem Fall werden 3-fach zum Training verwendet, 1-fach zur Validierung und die verbleibende Faltung zum Testen. Somit wird das vorgeschlagene Modell fünfmal trainiert und die Gesamtleistung des fünffachen berechnet.

Tabelle 1. Statistik der vier in dieser Studie verwendeten Datensätze.
2.2. Negative Datensatzkonstruktion
Um ein Modell zu trainieren, das die Klassifizierung von Promotor- und Nicht-Promotor-Sequenzen genau durchführen kann, müssen wir den negativen Satz (Nicht-Promotor-Sequenzen) sorgfältig auswählen. Dieser Punkt ist entscheidend, um ein Modell gut verallgemeinerbar zu machen und daher seine Präzision beizubehalten, wenn es an anspruchsvolleren Datensätzen ausgewertet wird. Frühere Arbeiten wie (Qian et al., 2018) konstruierten einen negativen Satz durch zufällige Auswahl von Fragmenten aus Genom-Nicht-Promotor-Regionen. Offensichtlich ist dieser Ansatz nicht völlig sinnvoll, da es keinen Schnittpunkt zwischen positiven und negativen Mengen gibt. Somit findet das Modell leicht grundlegende Funktionen, um die beiden Klassen zu trennen. Zum Beispiel kann das TATA-Motiv in allen positiven Sequenzen an einer bestimmten Position gefunden werden (normalerweise 28 bp stromaufwärts des TSS, zwischen –30 und –25 pb in unserem Datensatz). Wenn Sie also zufällig eine negative Menge erstellen, die dieses Motiv nicht enthält, wird in diesem Datensatz eine hohe Leistung erzielt. Das Modell kann jedoch negative Sequenzen mit TATA-Motiv nicht als Promotoren klassifizieren. Kurz gesagt, der Hauptfehler bei diesem Ansatz besteht darin, dass beim Trainieren eines Deep-Learning-Modells nur gelernt wird, die positiven und negativen Klassen anhand des Vorhandenseins oder Fehlens einiger einfacher Merkmale an bestimmten Positionen zu unterscheiden, was diese Modelle nicht praktikabel macht. In dieser Arbeit wollen wir dieses Problem lösen, indem wir eine alternative Methode etablieren, um die negative Menge von der positiven abzuleiten.
Unsere Methode basiert auf der Tatsache, dass immer dann, wenn die Merkmale zwischen der negativen und der negativen gemeinsam sind positive Klasse Das Modell neigt dazu, bei der Entscheidung seine Abhängigkeit von diesen Merkmalen zu ignorieren oder zu verringern (dh diesen Merkmalen niedrige Gewichte zuzuweisen). Stattdessen muss das Modell nach tieferen und weniger offensichtlichen Merkmalen suchen. Deep-Learning-Modelle leiden im Allgemeinen unter einer langsamen Konvergenz, während sie mit dieser Art von Daten trainieren. Diese Methode verbessert jedoch die Robustheit des Modells und stellt die Verallgemeinerung sicher. Wir rekonstruieren die negative Menge wie folgt. Jede positive Sequenz erzeugt eine negative Sequenz. Die positive Sequenz ist in 20 Teilsequenzen unterteilt. Dann werden 12 Teilsequenzen zufällig ausgewählt und zufällig ersetzt. Die verbleibenden 8 Teilsequenzen bleiben erhalten. Dieser Prozess ist in Abbildung 1 dargestellt. Die Anwendung dieses Prozesses auf den positiven Satz führt zu neuen Nicht-Promotor-Sequenzen mit konservierten Teilen aus Promotor-Sequenzen (unveränderte Teilsequenzen, 8 von 20 Teilsequenzen). Diese Parameter ermöglichen die Erzeugung eines negativen Satzes, dessen Sequenzen 32 und 40% konservierte Teile der Promotorsequenzen enthalten. Dieses Verhältnis hat sich als optimal erwiesen, um einen robusten Promotor-Prädiktor zu haben, wie in Abschnitt 3.2 erläutert.Da die konservierten Teile in den negativen Sequenzen die gleichen Positionen einnehmen, sind die offensichtlichen Motive wie TATA-Box und TSS nun zwischen den beiden Sätzen mit einem Verhältnis von 32 bis 40% gemeinsam. Die Sequenzlogos der positiven und negativen Sätze für die TATA-Promotordaten von Mensch und Maus sind in den 2 bzw. 3 gezeigt. Es ist ersichtlich, dass die positiven und negativen Sätze dieselben Grundmotive an denselben Positionen teilen, wie das TATA-Motiv an den Positionen -30 und –25 bp und das TSS an den Positionen +1 bp. Daher ist das Training anspruchsvoller, aber das resultierende Modell lässt sich gut verallgemeinern.
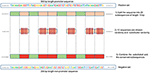
Abbildung 1. Darstellung der Negativsatzkonstruktionsmethode. Grün steht für die zufällig konservierten Teilsequenzen, während Rot für die zufällig ausgewählten und ersetzten Teilsequenzen steht.

Abbildung 2. Das Sequenzlogo im humanen TATA-Promotor sowohl für den positiven Satz (A) als auch für den negativen Satz (B). Die Diagramme zeigen die Erhaltung der Funktionsmotive zwischen den beiden Sätzen.

Abbildung 3. Das Sequenzlogo im Maus-TATA-Promotor sowohl für den positiven Satz (A) als auch für den negativen Satz (B). Die Diagramme zeigen die Erhaltung der Funktionsmotive zwischen den beiden Sätzen.
2.3. Die vorgeschlagenen Modelle
Wir schlagen ein Deep-Learning-Modell vor, das Faltungsschichten mit wiederkehrenden Schichten kombiniert, wie in Abbildung 4 gezeigt. Es akzeptiert eine einzelne genomische Rohsequenz, S = {N1, N2,…, Nl}, wobei N. ∈ {A, C, G, T} und l ist die Länge der Eingabesequenz als Eingabe und Ausgabe einer reellen Punktzahl. Der Eingang ist One-Hot-codiert und wird als eindimensionaler Vektor mit vier Kanälen dargestellt. Die Länge des Vektors l = 300 und der vier Kanäle sind A, C, G und T und werden als (1 0 0 0), (0 1 0 0), (0 0 1 0), (0 0 0 1) dargestellt ), beziehungsweise. Um das Modell mit der besten Leistung auszuwählen, haben wir die Rastersuchmethode zur Auswahl der besten Hyperparameter verwendet. Wir haben verschiedene Architekturen wie CNN allein, LSTM allein, BiLSTM allein, CNN in Kombination mit LSTM ausprobiert. Die eingestellten Hyperparameter sind die Anzahl der Faltungsschichten, die Kernelgröße, die Anzahl der Filter in jeder Schicht, die Größe der maximalen Pooling-Schicht, die Ausfallwahrscheinlichkeit und die Einheiten der Bi-LSTM-Schicht.
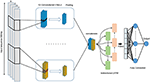
Abbildung 4. Die Architektur des vorgeschlagenen DeePromoter-Modells.
Das vorgeschlagene Modell beginnt mit mehreren Faltungsschichten, die parallel ausgerichtet sind und beim Erlernen der wichtigen Motive der Eingabesequenzen mit unterschiedlicher Fenstergröße helfen. Wir verwenden drei Faltungsschichten für Nicht-TATA-Promotoren mit Fenstergrößen von 27, 14 und 7 und zwei Faltungsschichten für TATA-Promotoren mit Fenstergrößen von 27, 14. Auf alle Faltungsschichten folgt die ReLU-Aktivierungsfunktion (Glorot et al. , 2011), eine maximale Pooling-Schicht mit einer Fenstergröße von 6 und eine Dropout-Schicht mit einer Wahrscheinlichkeit von 0,5. Dann werden die Ausgaben dieser Schichten miteinander verkettet und in eine bidirektionale Langzeit-Kurzzeitgedächtnisschicht (BiLSTM) (Schuster und Paliwal, 1997) mit 32 Knoten eingespeist, um die Abhängigkeiten zwischen den gelernten Motiven aus den Faltungsschichten zu erfassen. Die nach BiLSTM erlernten Merkmale werden abgeflacht, gefolgt von einem Ausfall mit einer Wahrscheinlichkeit von 0,5. Dann fügen wir zwei vollständig verbundene Schichten zur Klassifizierung hinzu. Der erste hat 128 Knoten, gefolgt von ReLU und Dropout mit einer Wahrscheinlichkeit von 0,5, während die zweite Schicht zur Vorhersage mit einem Knoten und einer Sigmoid-Aktivierungsfunktion verwendet wird. Mit BiLSTM können die Informationen bestehen bleiben und langfristige Abhängigkeiten von sequentiellen Proben wie DNA und RNA lernen. Dies wird durch die LSTM-Struktur erreicht, die aus einer Speicherzelle und drei Gattern besteht, die als Eingabe-, Ausgabe- und Vergessensgatter bezeichnet werden. Diese Gatter sind für die Regulierung der Informationen in der Speicherzelle verantwortlich. Darüber hinaus erhöht die Verwendung des LSTM-Moduls die Netzwerktiefe, während die Anzahl der erforderlichen Parameter gering bleibt. Ein tieferes Netzwerk ermöglicht das Extrahieren komplexerer Merkmale. Dies ist das Hauptziel unserer Modelle, da die negative Menge harte Stichproben enthält.
Das Keras-Framework wird zum Erstellen und Trainieren der vorgeschlagenen Modelle verwendet (Chollet F. et al., 2015). Der Adam-Optimierer (Kingma und Ba, 2014) wird zum Aktualisieren der Parameter mit einer Lernrate von 0,001 verwendet. Die Stapelgröße ist auf 32 und die Anzahl der Epochen auf 50 festgelegt. Ein vorzeitiges Stoppen wird basierend auf dem Validierungsverlust angewendet.
Ergebnisse und Diskussion
3.1. Leistungsmessungen
In dieser Arbeit verwenden wir die weit verbreiteten Bewertungsmetriken zur Bewertung der Leistung der vorgeschlagenen Modelle.Diese Metriken sind Präzision, Rückruf und Matthew-Korrelationskoeffizient (MCC) und werden wie folgt definiert:
Wenn TP wahr positiv ist und korrekt identifizierte Promotorsequenzen darstellt, TN wahr negativ ist und korrekt zurückgewiesene Promotorsequenzen darstellt, FP falsch positiv ist und falsch identifiziert darstellt Promotorsequenzen und FN ist falsch negativ und repräsentiert falsch abgelehnte Promotorsequenzen.
3.2. Auswirkung des Negativsatzes
Bei der Analyse der zuvor veröffentlichten Arbeiten zur Identifizierung von Promotorsequenzen haben wir festgestellt, dass die Leistung dieser Arbeiten stark von der Art der Erstellung des Negativdatensatzes abhängt. Sie zeigten eine sehr gute Leistung bei den von ihnen erstellten Datensätzen, weisen jedoch ein hohes falsch-positives Verhältnis auf, wenn sie an einem anspruchsvolleren Datensatz ausgewertet werden, der Nicht-Prompter-Sequenzen mit gemeinsamen Motiven mit Promotorsequenzen enthält. Zum Beispiel haben im Fall des TATA-Promotor-Datensatzes die zufällig erzeugten Sequenzen kein TATA-Motiv an den Positionen -30 und –25 bp, was wiederum die Aufgabe der Klassifizierung erleichtert. Mit anderen Worten, ihr Klassifikator hing vom Vorhandensein des TATA-Motivs ab, um die Promotorsequenz zu identifizieren, und als Ergebnis war es einfach, mit den von ihnen erstellten Datensätzen eine hohe Leistung zu erzielen. Ihre Modelle versagten jedoch dramatisch, wenn es um negative Sequenzen ging, die TATA-Motive enthielten (harte Beispiele). Die Genauigkeit nahm mit zunehmender Falsch-Positiv-Rate ab. Sie klassifizierten diese Sequenzen einfach als positive Promotorsequenzen. Eine ähnliche Analyse gilt für die anderen Promotormotive. Daher besteht der Hauptzweck unserer Arbeit nicht nur darin, eine hohe Leistung für einen bestimmten Datensatz zu erzielen, sondern auch die Modellfähigkeit bei der Verallgemeinerung durch Training an einem herausfordernden Datensatz zu verbessern.
Um diesen Punkt besser zu veranschaulichen, trainieren wir und Testen Sie unser Modell an TATA-Promotor-Datensätzen von Mensch und Maus mit verschiedenen Methoden zur Vorbereitung negativer Sätze. Das erste Experiment wird unter Verwendung zufällig abgetasteter negativer Sequenzen aus nicht-kodierenden Regionen des Genoms durchgeführt (d. H. Ähnlich dem in den vorherigen Arbeiten verwendeten Ansatz). Bemerkenswerterweise erreicht unser vorgeschlagenes Modell eine nahezu perfekte Vorhersagegenauigkeit (Präzision = 99%, Rückruf = 99%, Mcc = 98%) bzw. (Präzision = 99%, Rückruf = 98%, Mcc = 97%) für Mensch und Maus . Diese hohen Ergebnisse werden erwartet, aber die Frage ist, ob dieses Modell die gleiche Leistung beibehalten kann, wenn es an einem Datensatz ausgewertet wird, der harte Beispiele enthält. Die Antwort, basierend auf der Analyse der Vorgängermodelle, lautet Nein. Das zweite Experiment wird mit unserer vorgeschlagenen Methode zur Erstellung des Datensatzes durchgeführt, wie in Abschnitt 2.2 erläutert. Wir bereiten die Negativsätze vor, die eine konservierte TATA-Box mit unterschiedlichen Prozentsätzen wie 12, 20, 32 und 40% enthalten. Ziel ist es, die Lücke zwischen Präzision und Rückruf zu verringern. Dies stellt sicher, dass unser Modell komplexere Funktionen lernt, anstatt nur das Vorhandensein oder Fehlen einer TATA-Box zu lernen. Wie in 5A, B gezeigt, stabilisiert sich das Modell bei dem Verhältnis 32 ~ 40% sowohl für TATA-Promotor-Datensätze von Mensch als auch von Maus.
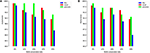
Abbildung 5. Die Auswirkung verschiedener Konservierungsverhältnisse des TATA-Motivs im Negativsatz auf die Leistung im Fall eines TATA-Promotor-Datensatzes sowohl für Mensch (A) als auch für Maus (B) .
3.3. Ergebnisse und Vergleich
In den letzten Jahren wurden zahlreiche Tools zur Vorhersage der Promotorregion vorgeschlagen (Hutchinson, 1996; Scherf et al., 2000; Reese, 2001; Umarov und Solovyev, 2017). Einige dieser Tools stehen jedoch nicht öffentlich zum Testen zur Verfügung, und einige von ihnen erfordern neben den genomischen Rohsequenzen weitere Informationen. In dieser Studie vergleichen wir die Leistung unserer vorgeschlagenen Modelle mit der aktuellen Arbeit auf dem neuesten Stand der Technik, CNNProm, die von Umarov und Solovyev (2017) vorgeschlagen wurde, wie in Tabelle 2 gezeigt. Im Allgemeinen sind die vorgeschlagenen Modelle DeePromoter, Übertreffen Sie CNNProm in allen Datensätzen mit allen Bewertungsmetriken deutlich. Insbesondere verbessert DeePromoter die Präzision, den Rückruf und das MCC im Fall eines menschlichen TATA-Datensatzes um 0,18, 0,04 bzw. 0,26. Im Fall eines menschlichen Nicht-TATA-Datensatzes verbessert DeePromoter die Genauigkeit um 0,39, den Rückruf um 0,12 und den MCC um 0,66. In ähnlicher Weise verbessert DeePromoter die Präzision und das MCC im Fall des Maus-TATA-Datensatzes um 0,24 bzw. 0,31. Im Fall eines Maus-Nicht-TATA-Datensatzes verbessert DeePromoter die Genauigkeit um 0,37, den Rückruf um 0,04 und das MCC um 0,65. Diese Ergebnisse bestätigen, dass CNNProm negative Sequenzen mit TATA-Promotor nicht abstößt, daher hat es einen hohen falsch positiven Wert. Andererseits sind unsere Modelle in der Lage, diese Fälle erfolgreicher zu behandeln, und die Falsch-Positiv-Rate ist im Vergleich zu CNNProm niedriger.

Tabelle 2. Vergleich des DeePromoter mit dem Status von -the-art-Methode.
Für weitere Analysen untersuchen wir die Wirkung alternierender Nukleotide an jeder Position auf den Output-Score. Wir konzentrieren uns auf die Region –40 und 10 bp, da sie den wichtigsten Teil der Promotorsequenz enthält. Für jede Promotorsequenz im Testsatz führen wir ein rechnergestütztes Mutationsscannen durch, um den Effekt der Mutation jeder Basis der Eingabesubsequenz zu bewerten (150 Substitutionen im Intervall –40 ~ 10 bp Subsequenz). Dies ist in den 6, 7 für TATA-Datensätze von Mensch und Maus dargestellt. Die blaue Farbe stellt einen Abfall der Ausgabewerte aufgrund einer Mutation dar, während die rote Farbe das Inkrement der Bewertung aufgrund einer Mutation darstellt. Wir stellen fest, dass die Änderung der Nukleotide zu C oder G in der Region –30 und –25 bp den Output-Score signifikant verringert. Diese Region ist eine TATA-Box, die ein sehr wichtiges funktionelles Motiv in der Promotorsequenz darstellt. So kann unser Modell die Bedeutung dieser Region erfolgreich erkennen. In den übrigen Positionen sind C- und G-Nukleotide gegenüber A und T vorzuziehen, insbesondere im Fall der Maus. Dies kann durch die Tatsache erklärt werden, dass die Promotorregion mehr C- und G-Nukleotide als A und T aufweist (Shi und Zhou, 2006).

Abbildung 6. Die Ausprägungskarte der Region –40 bp bis 10 bp, einschließlich der TATA-Box, bei humanen TATA-Promotorsequenzen.

Abbildung 7. Die Ausprägungskarte der Region –40 bp bis 10 bp, einschließlich der TATA-Box, im Fall von Maus-TATA-Promotorsequenzen.
Schlussfolgerung
Eine genaue Vorhersage der Promotorsequenzen ist wichtig, um den zugrunde liegenden Mechanismus des Genregulationsprozesses zu verstehen. In dieser Arbeit haben wir DeePromoter entwickelt, der auf einer Kombination aus Faltungs-Neuronales Netzwerk und bidirektionalem LSTM basiert, um die kurzen Eukaryoten-Promotorsequenzen bei Mensch und Maus sowohl für TATA- als auch für Nicht-TATA-Promotoren vorherzusagen. Die wesentliche Komponente dieser Arbeit bestand darin, das Problem der geringen Präzision (hohe Falsch-Positiv-Rate) zu überwinden, das bei den zuvor entwickelten Werkzeugen festgestellt wurde, da bei der Klassifizierung von Promotor- und Nicht-Promotor-Sequenzen einige offensichtliche Merkmale / Motive in der Sequenz verwendet wurden. In dieser Arbeit waren wir besonders daran interessiert, eine harte negative Menge zu konstruieren, die die Modelle dazu bringt, die Sequenz auf tiefe und relevante Merkmale zu untersuchen, anstatt nur die Promotor- und Nicht-Promotor-Sequenzen anhand der Existenz einiger funktioneller Motive zu unterscheiden. Der Hauptvorteil der Verwendung von DeePromoter besteht darin, dass die Anzahl falsch positiver Vorhersagen erheblich reduziert wird und gleichzeitig eine hohe Genauigkeit bei anspruchsvollen Datensätzen erzielt wird. DeePromoter übertraf die bisherige Methode nicht nur in Bezug auf die Leistung, sondern auch in Bezug auf die Überwindung des Problems hoher falsch positiver Vorhersagen. Es wird davon ausgegangen, dass dieses Framework in drogenbezogenen Anwendungen und im akademischen Bereich hilfreich sein könnte.
Autorenbeiträge
MO und ZL bereiteten den Datensatz vor, konzipierten den Algorithmus und führten das Experiment und durch Analyse. MO und HT haben den Webserver vorbereitet und das Manuskript mit Unterstützung von ZL und KC verfasst. Alle Autoren diskutierten die Ergebnisse und trugen zum endgültigen Manuskript bei.
Finanzierung
Diese Forschung wurde vom Gehirnforschungsprogramm der National Research Foundation (NRF) unterstützt, das von der koreanischen Regierung finanziert wurde ( MSIT) (Nr. NRF-2017M3C7A1044815).
Interessenkonflikterklärung
Die Autoren erklären, dass die Untersuchung ohne kommerzielle oder finanzielle Beziehungen durchgeführt wurde, die als ausgelegt werden könnten ein potenzieller Interessenkonflikt.
Bharanikumar, R., Premkumar, KAR und Palaniappan, A. (2018). Promotorpredict: Die sequenzbasierte Modellierung der Promotorstärke von Escherichia coli σ70 ergibt eine logarithmische Abhängigkeit zwischen Promotorstärke und Sequenz. PeerJ 6: e5862. doi: 10.7717 / peerj.5862
PubMed Abstract | CrossRef Volltext | Google Scholar
Glorot, X., Bordes, A. und Bengio, Y. (2011). „Neuronale Netze mit tiefem Gleichrichter“ in Proceedings of the Fourteenth International Conference on Artificial Intelligence and Statistics (Fort Lauderdale, FL 🙂 315–323.
Google Scholar
PubMed Abstract | Google Scholar
Kingma, DP und Ba, J. (2014). Adam: Eine Methode zur stochastischen Optimierung. arXiv-Vorabdruck arXiv: 1412.6980.
Google Scholar
Knudsen, S. (1999). Promotor2. 0: zur Erkennung von Polii-Promotorsequenzen. Bioinformatics 15, 356–361.
PubMed Abstract | Google Scholar
Ponger, L. und Mouchiroud, D. (2002). Cpgprod: Identifizierung von cpg-Inseln, die mit Transkriptionsstartstellen in großen genomischen Säugetiersequenzen assoziiert sind. Bioinformatics 18, 631–633. doi: 10.1093 / bioinformatics / 18.4.631
PubMed Abstract | CrossRef Volltext | Google Scholar
Quang, D. und Xie, X. (2016). Danq: Ein hybrides Faltungs- und wiederkehrendes tiefes neuronales Netzwerk zur Quantifizierung der Funktion von DNA-Sequenzen. Nucleic Acids Res. 44, e107 – e107. doi: 10.1093 / nar / gkw226
PubMed Abstract | CrossRef Volltext | Google Scholar
Umarov, R. K. und Solovyev, V. V. (2017). Erkennung von prokaryotischen und eukaryotischen Promotoren unter Verwendung von Faltungs-Deep-Learning-Neuronalen Netzen. PLoS ONE 12: e0171410. doi: 10.1371 / journal.pone.0171410
PubMed Abstract | CrossRef Volltext | Google Scholar