Határok a genetikában
Bevezetés
A promóterek azok a kulcselemek, amelyek a genom nem kódoló régióihoz tartoznak. Nagyrészt ellenőrzik a gének aktiválását vagy visszaszorítását. A gén transzkripciós kezdőhelye (TSS) közelében és felfelé helyezkednek el. A gén promóterének szomszédos régiója sok fontos (5 és 15 bázis hosszú) rövid DNS-elemet és motívumot tartalmazhat, amelyek felismerési helyként szolgálnak a fehérjék számára, amelyek a downstream gén transzkripciójának megfelelő iniciálása és szabályozása (Juven-Gershon et al., 2008). A gén transzkriptum megindítása a legalapvetőbb lépés a génexpresszió szabályozásában. A promóter mag egy minimális szakasza a DNS-szekvenciának, amely konszolidálja a TSS-t, és elegendő a transzkripció közvetlen elindításához. A mag promoter hossza általában 60 és 120 bázispár (bp) között van.
A TATA-box egy promóter szekvencia, amely más molekulák számára jelzi, hogy hol kezdődik a transzkripció. “TATA-box” névre keresztelték, mivel szekvenciáját a T és A bázispárok (TATAAA) ismétlése jellemzi (Baker et al., 2003). A TATA-boxon végzett vizsgálatok túlnyomó részét emberi, élesztő, és Drosophila genomok, azonban hasonló elemeket találtak más fajokban is, mint az archaea és az ősi eukarióták (Smale és Kadonaga, 2003). Emberi esetben a gének 24% -ában vannak TATA-boxot tartalmazó promoter régiók (Yang et al., 2007 Az eukariótákban a TATA-box ~ 25 bp-nél helyezkedik el a TSS előtt (Xu és mtsai., 2016). Képes meghatározni a transzkripció irányát, és jelzi az elolvasandó DNS-szálat is. számos nem kódoló régióhoz kötődik, beleértve a TATA-boxot, és toboroz egy RNS-polimeráz nevű enzimet, amely a DNS-ből szintetizálja az RNS-t.
A promóterek fontos szerepe miatt a génátírásban a promóter helyek pontos előrejelzése válik szükséges lépés a génexpresszióban, a minták értelmezésében, valamint az építésben és a megértésben genetikai szabályozó hálózatok funkcionalitását. Különböző biológiai kísérletek voltak a promóterek azonosítására, mint például a mutációs elemzés (Matsumine et al., 1998) és az immunprecipitációs vizsgálatok (Kim és mtsai, 2004; Dahl és Collas, 2008). Ezek a módszerek azonban drágák és időigényesek is voltak. A közelmúltban a következő generációs szekvenálás (NGS) (Behjati és Tarpey, 2013) kifejlesztésével több különböző organizmus génjét szekvenálták, és génelemeiket számítási szempontból feltárták (Zhang et al., 2011). Másrészt az NGS technológia innovációja a teljes genomszekvenálás költségeinek drámai csökkenését eredményezte, így több szekvenálási adat áll rendelkezésre. Az adatok elérhetősége vonzza a kutatókat arra, hogy számítási modelleket dolgozzanak ki a promóter előrejelzési feladatához. Ez azonban még mindig hiányos feladat, és nincs hatékony szoftver, amely pontosan meg tudná jósolni a promótereket. és a GpG-alapú megközelítés. A jelalapú prediktorok az RNS polimeráz kötőhelyhez kapcsolódó promóter elemekre összpontosítanak, és figyelmen kívül hagyják a szekvencia nem elem részeit. Ennek eredményeként az előrejelzési pontosság gyenge volt, és nem volt kielégítő. A jelalapú prediktorok példái a következők: PromoterScan (Prestridge, 1995), amely a TATA-box kivont jellemzőit és a transzkripciós faktor-kötőhelyek súlyozott mátrixát lineáris diszkriminátorral használta a nem-promóteres promóter-szekvenciák osztályozására; A Promoter2.0 (Knudsen, 1999), amely különféle dobozokból (például TATA-Box, CAAT-Box és GC-Box) vonta ki a jellemzőket, és osztályozás céljából továbbította azokat a mesterséges neurális hálózatoknak (ANN) Az NNPP2.1 (Reese, 2001), amely az iniciátor elemet (Inr) és a TATA-Box-ot használta a funkciók kinyerésére, és az idõ késleltetésû neurális hálózatot az osztályozáshoz, valamint Down és Hubbard (2002), amelyek a TATA-Box-ot használták, és releváns vektor-gépeket használtak (RVM) osztályozóként. A tartalomalapú prediktorok a k-mer gyakoriságának megszámlálására támaszkodtak, k-hosszú ablak futtatásával a szekvencián. Ezek a módszerek azonban figyelmen kívül hagyják a szekvenciák bázispárjainak térinformációját. A tartalomalapú prediktorok példái a következők: PromFind (Hutchinson, 1996), amely a k-mer frekvenciát használta a hexamer promóter előrejelzéséhez; PromoterInspector (Scherf és mtsai, 2000), amely a promotereket tartalmazó régiókat azonosította a polimeráz II promoterek közös genomiális kontextusa alapján, változó hosszúságú motívumokként meghatározott specifikus jellemzők beolvasásával; MCPromoter1.1 (Ohler és mtsai, 1999), amely egyetlen promóter szekvencia előrejelzésére az 5. rendű egyetlen interpolált Markov-láncot (IMC) használta.Végül a GpG-alapú prediktorok a GpG-szigetek elhelyezkedését használták fel, mivel a promóter régió vagy az emberi gének első exon régiója általában GpG-szigeteket tartalmaz (Ioshikhes és Zhang, 2000; Davuluri és mtsai, 2001; Lander és mtsai, 2001; Ponger és Mouchiroud, 2002). A promóterek csupán 60% -a tartalmaz GpG-szigeteket, ezért az ilyen típusú prediktorok predikciós pontossága soha nem haladta meg a 60% -ot.
A közelmúltban szekvencia alapú megközelítéseket alkalmaztak a promóter predikciójához. Yang és mtsai. (2017) különböző jellemzőkivonási stratégiákat használt fel a legrelevánsabb szekvenciainformációk megfogására annak érdekében, hogy megjósolja az enhancer-promoter interakciókat. Lin és mtsai. (2017) egy szekvencia alapú prediktort, “iPro70-PseZNC” elnevezést javasolt a sigma70 promoter azonosítására a prokariótában. Hasonlóképpen, Bharanikumar et al. (2018) javasolta a PromoterPredict-et az Escherichia coli promoterek erősségének előrejelzésére egy dinamikus többszörös regressziós megközelítés alapján, ahol a szekvenciákat pozíciós súlymátrixként (PWM) ábrázolták. Kanhere és Bansal (2005) a promóter és a nem promóter szekvenciák DNS-szekvencia stabilitásának különbségeit használta fel azok megkülönböztetésére. Xiao és mtsai. (2018) egy kétrétegű prediktort, az iPSW (2L) -PseKNC-t vezetett be a promóter szekvenciák azonosításához, valamint a promóterek erősségéhez a szekvenciák hibrid tulajdonságainak kivonásával.
Az összes fent említett prediktor domént igényel ismereteket a sajátosságok kézi készítéséhez. Másrészt a mély tanuláson alapuló megközelítések hatékonyabb modellek felépítését teszik lehetővé a nyers adatok (DNS / RNS szekvenciák) közvetlen felhasználásával. A mély konvolúciós ideghálózat a legmodernebb eredményeket érte el olyan kihívást jelentő feladatokban, mint a kép, videó, hang és beszéd feldolgozása (Krizhevsky et al., 2012; LeCun et al., 2015; Schmidhuber, 2015; Szegedy et al. , 2015). Ezenkívül sikeresen alkalmazták olyan biológiai problémákban is, mint a DeepBind (Alipanahi et al., 2015), DeepCpG (Angermueller et al., 2017), az elágazási pontok kiválasztása (Nazari et al., 2018), az alternatív splicing helyek előrejelzése (Oubounyt et al., 2018), 2 “-metilezési helyek előrejelzése (Tahir et al., 2018), DNS-szekvencia kvantifikáció (Quang és Xie, 2016), emberi fehérje szubcelluláris lokalizációja (Wei et al., 2018), stb. A CNN nemrégiben figyelemre méltó szerepet kapott a promóter-felismerési feladatban. Umarov és Solovyev (2017) nemrégiben bevezette a CNNprom-ot a rövid promóter-szekvenciák megkülönböztetésére. Ez a CNN-alapú architektúra magas eredményeket ért el a promoter és a nem promóter szekvenciák osztályozásában. Qian és munkatársai (2018), ahol a szerzők támogató vektor gép (SVM) osztályozót alkalmaztak a legfontosabb promóter szekvencia elemek ellenőrzésére, majd a legbefolyásosabb elemeket tömörítetlenül tartották, miközben a kevésbé fontosakat tömörítették. Ez a folyamat jobb teljesítményt eredményezett. A közelmúltban hosszú promóter-azonosítási modellt javasoltak Umarov és munkatársai. (2019), amelyben a szerzők a TSS-pozíció azonosítására összpontosítottak.
Az összes fent említett munkában a negatív halmazt a genom nem promóteres régióiból vették ki. Annak tudatában, hogy a promóter szekvenciák kizárólag olyan specifikus funkcionális elemekben gazdagok, mint a TATA-box, amely –30 ~ –25 bp, GC-Box, amely –110 ~ –80 bp, CAAT-Box, amely a következő helyen található: 80 ~ –70 bp, stb. Ez nagy osztályozási pontosságot eredményez a pozitív és negatív minták szekvenciaszerkezetét tekintve óriási különbségek miatt. Ezenkívül az osztályozási feladat megkönnyíti az elérését, például a CNN modellek csak bizonyos motívumok jelenlétére vagy hiányára támaszkodnak a saját pozíciójukban a szekvencia típusának meghozatalakor. Így ezeknek a modelleknek nagyon alacsony a pontossága / érzékenysége (magas hamis pozitív), ha azokat olyan promóciós motívumokkal rendelkező genomi szekvenciákon tesztelik, amelyek nem promóter szekvenciák. Köztudott, hogy a genomban több TATAAA motívum található, mint amelyek a promóter régiókhoz tartoznak. Például önmagában az 1. emberi kromoszóma DNS-szekvenciája (ftp://ftp.ensembl.org/pub/release-57/fasta/homo_sapiens/dna/) 151 656 TATAAA motívumot tartalmaz. Több, mint a teljes emberi genomban a hozzávetőleges maximális gének száma. Ennek a kérdésnek a szemléltetéseként azt vesszük észre, hogy amikor ezeket a modelleket nem promóter szekvenciákon teszteljük, amelyeknek TATA doboza van, akkor ezeknek a szekvenciáknak a nagy részét rosszul osztályozzák. Ezért egy robusztus osztályozó létrehozásához a negatív halmazt gondosan kell kiválasztani, mivel meghatározza azokat a jellemzőket, amelyeket az osztályozó használni fog az osztályok megkülönböztetése érdekében. Ennek az ötletnek a fontosságát már korábbi munkák is bizonyították, például (Wei et al., 2014). Ebben a munkában főleg ezzel a kérdéssel foglalkozunk, és javaslatot teszünk egy olyan megközelítésre, amely egyesíti a negatív osztály pozitív osztályának funkcionális motívumait, hogy csökkentse a modell ezen motívumoktól való függését.Az LSTM modellel kombinált CNN-t használjuk az emberi és egér TATA és nem TATA eukarióta promóterek szekvenciajellemzőinek elemzéséhez, és olyan számítási modelleket építünk, amelyek pontosan megkülönböztethetik a rövid promóter szekvenciákat a nem promóterektől.
Anyagok és módszerek
2.1. Adatkészlet
Azokat az adathalmazokat, amelyeket a javasolt promóter prediktor képzésére és tesztelésére használnak, emberről és egérről gyűjtik össze. A promóterek két megkülönböztető osztályát tartalmazzák, nevezetesen a TATA promotereket (azaz a TATA-boxot tartalmazó szekvenciákat) és a nem TATA promótereket. Ezek az adatkészletek az Eukarióta Promoter Adatbázisból (EPDnew) épültek (Dreos et al., 2012). Az EPDnew egy új szakasz a jól ismert EPD adatkészlet alatt (Périer et al., 2000), amely az eukarióta POL II promoterek nem redundáns gyűjteményét jegyzi fel, ahol kísérletileg meghatározták a transzkripció kezdőhelyét. Kiváló minőségű promótereket biztosít az ENSEMBL promótergyűjteményhez képest (Dreos et al., 2012), és nyilvánosan elérhető a https://epd.epfl.ch//index.php címen. Az egyes szervezetekhez TATA és nem TATA promoter genom szekvenciákat töltöttünk le az EPDnew-ből. Ez a művelet négy promóter-adatkészlet megszerzését eredményezte, nevezetesen: Human-TATA, Human-non-TATA, Mouse-TATA és Mouse-non-TATA. Ezen adatkészletek mindegyikéhez egy negatív halmazt (nem promóter szekvenciákat) állítanak elő, amelyek ugyanolyan méretűek, mint a pozitív, a következő megközelítés alapján javasolt megközelítés alapján. Az egyes organizmusok promoter szekvenciáinak számát az 1. táblázat tartalmazza. Az összes szekvencia hossza 300 bp, és ezeket -249 ~ + 50 bp-ből extraháltuk (+1 a TSS-pozícióra vonatkozik). Minőségellenőrzésként ötszörös keresztellenőrzést alkalmaztunk a javasolt modell értékeléséhez. Ebben az esetben 3-szorosokat használnak az edzéshez, 1-szereseket az érvényesítéshez, a fennmaradó részt pedig a teszteléshez. Így a javasolt modellt ötször képezik ki, és kiszámítják az ötszörös teljes teljesítményét.

1. táblázat: A tanulmányban használt négy adatállomány statisztikája.
2.2. Negatív adatkészlet felépítés
Annak érdekében, hogy olyan modellt képezzünk, amely pontosan képes végrehajtani a promoter és a nem promóter szekvenciák osztályozását, gondosan meg kell választanunk a negatív halmazt (nem promóter szekvenciákat). Ez a pont döntő fontosságú annak érdekében, hogy a modell képes legyen általánosítani, és ezért meg tudja őrizni a pontosságát, ha nagyobb kihívást jelentő adatkészleteken értékelik. Korábbi munkák, például (Qian és mtsai, 2018), negatív halmazot konstruáltak úgy, hogy véletlenszerűen választottak ki fragmenseket a genom nem promóter régióiból. Nyilvánvaló, hogy ez a megközelítés nem teljesen ésszerű, mert ha nincs kereszteződés a pozitív és a negatív halmazok között. Így a modell könnyen megtalálja az alapvető jellemzőket a két osztály szétválasztására. Például a TATA motívum minden pozitív szekvenciában megtalálható egy adott helyzetben (általában 28 bp-tal a TSS előtt, az adatkészletünkben –30 és –25 pb között). Ezért véletlenszerűen negatív halmaz létrehozása, amely nem tartalmazza ezt a motívumot, nagy teljesítményt fog eredményezni ebben az adatkészletben. A modell azonban nem képes negatív szekvenciákat, amelyek TATA motívummal rendelkeznek, promóterként osztályozni. Röviden, ennek a megközelítésnek a legfőbb hibája az, hogy a mély tanulási modell képzésénél csak a pozitív és a negatív osztályok megkülönböztetését tanulja meg bizonyos egyszerű tulajdonságok megléte vagy hiánya alapján meghatározott pozíciókban, ami ezeket a modelleket kivitelezhetetlenné teszi. Ebben a munkában arra a célra törekszünk, hogy alternatív módszert hozzunk létre a negatív halmaz pozitívból való levezetésére.
Módszerünk azon a tényen alapul, hogy amikor a jellemzők közösek a negatív és a negatív között pozitív osztály, a modell a döntés meghozatalakor hajlamos figyelmen kívül hagyni vagy csökkenteni függését ezektől a tulajdonságoktól (azaz alacsony súlyokat rendel ezekhez a tulajdonságokhoz). Ehelyett a modell kénytelen mélyebb és kevésbé nyilvánvaló jellemzőket keresni. A mély tanulási modellek általában lassú konvergenciában szenvednek, miközben ilyen típusú adatokra készülnek. Ez a módszer azonban javítja a modell robusztusságát és biztosítja az általánosítást. A negatív halmazt az alábbiak szerint rekonstruáljuk. Minden pozitív szekvencia egy negatív szekvenciát generál. A pozitív szekvenciát 20 részre osztjuk. Ezután 12 szekvenciát véletlenszerűen kiválasztunk és véletlenszerűen helyettesítünk. A fennmaradó 8 szekvenciát konzerváltuk. Ezt a folyamatot az 1. ábra szemlélteti. Ezt a folyamatot alkalmazva a pozitív halmaz eredményeként új, nem promóter szekvenciákban, promóter szekvenciák konzervált részeivel (változatlan szekvenciák, 8 szekvencia 20-ból). Ezek a paraméterek lehetővé teszik egy olyan negatív halmaz előállítását, amelynek szekvenciái 32 és 40% -ban tartalmazzák a promóter szekvenciák konzervált részeit. Ezt az arányt optimálisnak találják a robusztus promóter prediktor használatához, amint azt a 3.2 szakasz leírja.Mivel a konzervált részek ugyanazt a pozíciót foglalják el a negatív szekvenciákban, a nyilvánvaló motívumok, mint például a TATA-box és a TSS, most már közösek a két halmaz között 32 ~ 40% arányban. A pozitív és a negatív halmaz szekvencia logóit mind az emberi, mind az egér TATA promoter adatokhoz a 2., 3. ábra mutatja. Látható, hogy a pozitív és a negatív halmaz ugyanazon az alapmotívumon osztozik ugyanabban a helyzetben, például a TATA-motívum a -30 és –25 bp pozícióban, a TSS pedig a +1 bp pozícióban. Ezért a képzés nagyobb kihívást jelent, de a kapott modell jól általánosít.
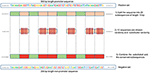
1. ábra: A negatív halmazkonstrukciós módszer illusztrációja. A zöld a véletlenszerűen konzervált részeket, míg a piros a véletlenszerűen kiválasztott és helyettesített részeket jelenti.

2. ábra: A szekvencia logója az emberi TATA promóterben pozitív (A) és negatív halmaz (B) esetén egyaránt. A diagramok a két halmaz közötti funkcionális motívumok megőrzését mutatják.

3. ábra: A szekvencia embléma az egér TATA promóterében pozitív (A) és negatív halmaz (B) esetén egyaránt. A diagramok a két halmaz közötti funkcionális motívumok megőrzését mutatják be.
2.3. A javasolt modellek
Javasoljuk egy mély tanulási modellt, amely a konvolúciós rétegeket és a visszatérő rétegeket ötvözi a 4. ábrán látható módon. Egyetlen nyers genomi szekvenciát fogad el, S = {N1, N2,…, Nl}, ahol N ∈ {A, C, G, T} és l a bemeneti szekvencia hossza, bemenetként és kimenetként valós értékű pontszám. A bemenet egy forró kódolású és négy csatornás egydimenziós vektorként van ábrázolva. Az l = 300 vektor és a négy csatorna hossza A, C, G és T, és (1 0 0 0), (0 1 0 0), (0 0 1 0), (0 0 0 1) ), ill. A legjobban teljesítő modell kiválasztásához rácskeresési módszert használtunk a legjobb hiperparaméterek kiválasztásához. Különböző architektúrákat próbáltunk ki, mint például egyedül a CNN, egyedül az LSTM, egyedül a BiLSTM, az LSTM-mel kombinált CNN. A hangolt hiperparaméterek a konvolúciós rétegek száma, a kernel mérete, az egyes rétegekben lévő szűrők száma, a maximális pooling réteg mérete, a lemorzsolódás valószínűsége és a Bi-LSTM réteg egységei.
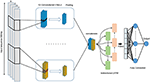
4. ábra A javasolt DeePromoter modell architektúrája.
A javasolt modell több konvolúciós réteggel indul, amelyek párhuzamosan vannak egymáshoz igazítva, és segítenek megismerni a különböző ablakméretű bemeneti szekvenciák fontos motívumait. Három konvolúciós réteget használunk nem TATA promóterhez, 27, 14 és 7 ablakmérettel, és két konvolúciós réteget a 27, 14 ablakméretű TATA promóterekhez. Az összes konvolúciós réteget ReLU aktivációs funkció követi (Glorot et al. , 2011), egy maximális pooling réteg, amelynek ablakmérete 6, és egy dropout réteg, amelynek valószínűsége 0,5. Ezután ezeknek a rétegeknek a kimenetét összefűzzük, és egy kétirányú hosszú távú memória (BiLSTM) (Schuster és Paliwal, 1997) 32 csomópontú rétegbe tápláljuk a konvolúciós rétegek tanult motívumai közötti függőségek megragadása érdekében. A BiLSTM után elsajátított jellemzőket ellapítjuk, majd 0,5-es valószínűséggel lemorzsolódást követünk. Ezután két teljesen összekapcsolt réteget adunk az osztályozáshoz. Az elsőnek 128 csomópontja van, ezt követi a ReLU és a lemorzsolódás 0,5 valószínűséggel, míg a második réteget egy csomópontos és szigmoid aktivációs funkcióval történő predikcióhoz használják. A BiLSTM lehetővé teszi az információk fennmaradását és a szekvenciális minták, például a DNS és az RNS hosszú távú függőségeinek megismerését. Ez az LSTM struktúrán keresztül érhető el, amely egy memória cellából és három kapuból áll, amelyeket bemeneti, kimeneti és elfelejtett kapuknak neveznek. Ezek a kapuk felelősek a memória cellában lévő információk szabályozásáért. Ezenkívül az LSTM modul használata növeli a hálózat mélységét, miközben a szükséges paraméterek száma továbbra is alacsony. A mélyebb hálózat lehetővé teszi a bonyolultabb funkciók kinyerését, és ez a modelljeink fő célja, mivel a negatív halmaz kemény mintákat tartalmaz.
A Keras keretrendszert használják a javasolt modellek felépítésére és képzésére (Chollet F. et al., 2015). Az Adam optimalizáló (Kingma és Ba, 2014) a paraméterek frissítésére szolgál 0,001 tanulási sebességgel. A kötegelt méret 32-re, a korszakok száma pedig 50-re van állítva. A korai leállítást az érvényesítési veszteség alapján alkalmazzák.
Eredmények és megbeszélés
3.1. Teljesítménymérők
Ebben a munkában a széles körben elfogadott értékelési mutatókat használjuk a javasolt modellek teljesítményének értékeléséhez.Ezek a mutatók a pontosság, a visszahívás és a Matthew korrelációs együttható (MCC), és a következőképpen vannak meghatározva:
Ahol a TP valóban pozitív és helyesen azonosított promóter szekvenciákat képvisel, a TN igaz negatív és helyesen elutasított promóter szekvenciákat képvisel, az FP hamis pozitív és helytelenül azonosítottakat képvisel promóter szekvenciák, az FN pedig hamis negatív és helytelenül elutasított promóter szekvenciákat képvisel.
3.2. A negatív halmaz hatása
A korábban publikált művek promóter szekvenciák azonosításának elemzése során észrevettük, hogy e művek teljesítménye nagymértékben függ a negatív adatkészlet elkészítésének módjától. Nagyon jól teljesítettek az általuk készített adatkészleteken, azonban magas hamis pozitív arányuk van, ha egy nagyobb kihívást jelentő adatkészleten értékelték, amely nem promóter szekvenciákat tartalmaz, amelyek közös motívumokkal rendelkeznek a promóter szekvenciákkal. Például a TATA promoter adatkészlet esetén a véletlenszerűen generált szekvenciáknak nem lesz TATA motívuma a -30 és –25 bp pozícióban, ami viszont megkönnyíti a besorolást. Más szóval, osztályozójuk a TATA motívum jelenlététől függött a promóter szekvencia azonosításához, és ennek eredményeként könnyű volt magas teljesítményt elérni az általuk készített adatkészleteken. Modelljeik azonban drámai kudarcot vallottak, amikor negatív szekvenciákkal foglalkoztak, amelyek TATA motívumot tartalmaztak (kemény példák). A pontosság a hamis pozitív arány növekedésével csökkent. Egyszerűen ezeket a szekvenciákat pozitív promóter szekvenciáknak minősítették. Hasonló elemzés érvényes a többi promoter motívumra is. Ezért munkánk fő célja nemcsak egy adott adatkészlet magas teljesítményének elérése, hanem a modell általános képességének javítása is az általánosítás terén egy kihívást jelentő adatkészleten történő képzéssel.
Ennek a pontnak a további szemléltetése érdekében edzünk és teszteljük modellünket az emberi és egér TATA promoter adatkészleteken, negatív halmazok előállításának különböző módszereivel. Az első kísérletet véletlenszerűen mintavételezett negatív szekvenciák felhasználásával hajtjuk végre a genom nem kódoló régióiból (vagyis hasonlóan az előző munkákban alkalmazott megközelítéshez). Figyelemre méltó, hogy javasolt modellünk szinte tökéletes predikciós pontosságot (precízió = 99%, visszahívás = 99%, Mcc = 98%) és (pontosság = 99%, visszahívás = 98%, Mcc = 97%) ér el mind emberi, mind egér esetében . Ezek a magas eredmények várhatóak, de az a kérdés, hogy ez a modell képes-e fenntartani ugyanazt a teljesítményt, ha egy kemény példákkal rendelkező adatkészleten értékelik. A korábbi modellek elemzése alapján a válasz nemleges. A második kísérletet az adatkészlet elkészítéséhez javasolt módszerünkkel hajtjuk végre, a 2.2 szakaszban leírtak szerint. Előkészítjük azokat a negatív halmazokat, amelyek konzervált TATA-boxot tartalmaznak, különböző százalékokkal, például 12, 20, 32 és 40%, és a cél a pontosság és a visszahívás közötti szakadék csökkentése. Ez biztosítja, hogy modellünk bonyolultabb funkciókat tanuljon meg, ne csak a TATA-box jelenlétét vagy hiányát tanulja meg. Amint az 5A, B ábrákon látható, a modell 32 ~ 40% -os arányban stabilizálódik mind az emberi, mind az egér TATA promoter adatkészleteknél.
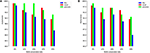
5. ábra A negatív halmazban a TATA motívum különböző megőrzési arányainak hatása a teljesítményre a TATA promoter adathalmaz esetében mind az ember (A), mind az egér (B) esetében .
3.3. Eredmények és összehasonlítás
Az elmúlt években rengeteg promóter régió előrejelzési eszközt javasoltak (Hutchinson, 1996; Scherf és mtsai, 2000; Reese, 2001; Umarov és Solovyev, 2017). Ezen eszközök egy része azonban nyilvánosan nem áll rendelkezésre tesztelésre, és néhányuk a nyers genomi szekvenciákon kívül további információkra szorul. Ebben a tanulmányban összehasonlítjuk a javasolt modellek teljesítményét az aktuális, CNNProm munkával, amelyet Umarov és Solovyev (2017) javasolt, a 2. táblázat szerint. Általában a javasolt modellek, a DeePromoter, egyértelműen felülmúlja a CNNProm-ot minden adatkészletben, minden értékelési mutatóval. Pontosabban, a DeePromoter 0,18, 0,04 és 0,26-kal javítja a pontosságot, a visszahívást és az MCC-t az emberi TATA-adatkészlet esetében. Emberi nem TATA adatkészlet esetén a DeePromoter 0,39-rel, a visszahívás 0,12-rel, az MCC pedig 0,66-tal javítja a pontosságot. Hasonlóképpen, a DeePromoter javítja az egér TATA adatkészletének pontosságát, az MCC pedig 0,24, illetve 0,31. Az egér nem TATA adatkészlete esetén a DeePromoter 0,37-rel, a visszahívás 0,04-gyel, az MCC pedig 0,65-tel javítja a pontosságot. Ezek az eredmények megerősítik, hogy a CNNProm nem utasítja el a negatív szekvenciákat a TATA promóterrel, ezért magas hamis pozitív. Másrészt modelljeink sikeresebben képesek kezelni ezeket az eseteket, és a hamis pozitív arány alacsonyabb a CNNProm-hoz képest.

2. táblázat: A DeePromoter összehasonlítása az állapotával -a művészeti módszer.
További elemzésekhez megvizsgáljuk az egyes pozíciókban váltakozó nukleotidok hatását a kimeneti pontszámra. A –40 és 10 bp-os régióra koncentrálunk, mivel ez tartalmazza a promóter szekvencia legfontosabb részét. A tesztkészlet minden promóter-szekvenciájához számítógépes mutációs pásztázást végzünk, hogy értékeljük a bemeneti szekvencia minden bázisának mutációjának hatását (150 szubsztitúció a –40 ~ 10 bp szekvencián). Ezt a 6., 7. ábra szemlélteti az emberi, illetve az egér TATA adatkészleteivel. A kék szín a kimeneti pontszám csökkenését jelenti a mutáció miatt, míg a piros a mutáció miatti pontszám növekedését jelenti. Észrevesszük, hogy ha a nukleotidokat C-re vagy G-re változtatjuk, a –30 és –25 bp régióban jelentősen csökken a kimeneti pontszám. Ez a régió a TATA-box, amely nagyon fontos funkcionális motívum a promóter szekvenciában. Így modellünk sikeresen megtalálja ennek a régiónak a fontosságát. A többi helyzetben a C és G nukleotidok előnyösebbek, mint az A és T, különösen az egér esetében. Ez azzal magyarázható, hogy a promóter régióban több C és G nukleotid van, mint A és T (Shi és Zhou, 2006).

6. ábra – A TATA-dobozt tartalmazó –40 bp-től 10 bp-ig terjedő régió sűrűség térképe, humán TATA promoter szekvenciák esetén.

7. ábra. Egér TATA promóter-szekvenciák esetén a TATA-mezőt tartalmazó régió -40 és 10 bp közötti sűrűségtérkép.
Következtetés
A promóter szekvenciák pontos előrejelzése elengedhetetlen a génszabályozási folyamat mögöttes mechanizmusának megértéséhez. Ebben a munkában kifejlesztettük a DeePromoter -t, amely a konvolúciós ideghálózat és a kétirányú LSTM kombinációján alapul, hogy megjósolja a rövid eukarióta promoterszekvenciákat ember és egér esetén mind TATA, mind nem TATA promoter számára. E munka lényeges eleme az volt, hogy a promóteres és a nem-promóteres szekvenciák osztályozásakor a korábban kifejlesztett eszközökben észlelt alacsony pontosság (magas hamis pozitív arány) problémáját leküzdjék, mivel a szekvenciában néhány nyilvánvaló tulajdonságra / motívumra támaszkodtak. Ebben a munkában különösképpen egy kemény negatív halmaz felállítására voltunk kíváncsiak, amely a modelleket a mély és releváns jellemzők szekvenciájának feltárása felé készteti, ahelyett, hogy csak a funkcionális motívumok megléte alapján különböztetnénk meg a promóter és a nem promóter szekvenciákat. A DeePromoter használatának fő előnye, hogy jelentősen csökkenti a hamis pozitív előrejelzések számát, miközben nagy pontosságot ér el a kihívást jelentő adatkészleteknél. A DeePromoter nemcsak a teljesítményben, hanem a magas hamis pozitív előrejelzések kérdésének legyőzésében is felülmúlta az előző módszert. Az előrejelzések szerint ez a keretrendszer hasznos lehet a kábítószerrel kapcsolatos alkalmazásokban és az egyetemeken.
Szerzői közreműködések
MO és ZL elkészítette az adatkészletet, elkészítette az algoritmust, elvégezte a kísérletet és elemzés. MO és HT elkészítette a webszervert, és a ZL és a KC támogatásával megírta a kéziratot. Valamennyi szerző megvitatta az eredményeket, és hozzájárult az utolsó kézirat elkészítéséhez.
Finanszírozás
Ezt a kutatást a koreai kormány által finanszírozott Nemzeti Kutatási Alapítvány (NRF) Agykutatási Programja támogatta ( MSIT) (NRF-2017M3C7A1044815 sz.).
Összeférhetetlenségi nyilatkozat
A szerzők kijelentik, hogy a kutatást olyan kereskedelmi vagy pénzügyi kapcsolatok hiányában végezték, amelyek értelmezhetők potenciális összeférhetetlenség.
Bharanikumar, R., Premkumar, KAR, és Palaniappan, A. (2018). Promoterprediktum: az Escherichia coli σ70 promoter erősségének szekvencia alapú modellezése logaritmikus függőséget eredményez a promóter erőssége és a szekvencia között. PeerJ 6: e5862. doi: 10.7717 / peerj.5862
PubMed Abstract | CrossRef teljes szöveg | Google Tudós
Glorot, X., Bordes, A. és Bengio, Y. (2011). “Mély ritkán egyenirányító neurális hálózatok”, a Mesterséges Intelligencia és Statisztika Tizennegyedik Nemzetközi Konferenciájának közleményében (Fort Lauderdale, FL:) 315–333.
Google Scholar
Hutchinson, G. (1996). A gerinces promóter régiók előrejelzése differenciális hexamer frekvencia-analízissel. Bioinformatika 12, 391–398.
PubMed Abstract | Google Scholar
Kingma, DP és Ba, J. (2014). Adam: módszer sztochasztikus optimalizálásra. arXiv preprint arXiv: 1412.6980.
Google Scholar
Knudsen, S. (1999). Promoter2. 0: a polii promoter szekvenciák felismerésére. Bioinformatika 15, 356–361.
PubMed absztrakt | Google Tudós
Ponger, L. és Mouchiroud, D. (2002). Cpgprod: a transzkripciós starthelyekhez kapcsolódó cpg-szigetek azonosítása nagy genomi emlősszekvenciákban. Bioinformatika 18, 631–633. doi: 10.1093 / bioinformatics / 18.4.631
PubMed Abstract | CrossRef teljes szöveg | Google Tudós
Quang, D. és Xie, X. (2016). Danq: hibrid konvolúciós és visszatérő mély neurális hálózat a dna szekvenciák működésének számszerűsítésére. Nukleinsavak Res. 44., e107 – e107. doi: 10.1093 / nar / gkw226
PubMed Abstract | CrossRef teljes szöveg | Google Tudós
Umarov, R. K. és Solovyev, V. V. (2017). A prokarióta és eukarióta promóterek felismerése konvolúciós mély tanulási idegi hálózatok segítségével. PLoS ONE 12: e0171410. doi: 10.1371 / journal.pone.0171410
PubMed Abstract | CrossRef teljes szöveg | Google Tudós