Frontiere della genetica
Introduzione
I promotori sono gli elementi chiave che appartengono alle regioni non codificanti del genoma. Controllano in gran parte l’attivazione o la repressione dei geni. Si trovano vicino ea monte del sito di inizio della trascrizione (TSS) del gene. La regione fiancheggiante del promotore di un gene può contenere molti elementi e motivi di DNA corti cruciali (lunghi 5 e 15 basi) che servono come siti di riconoscimento per le proteine che forniscono corretto inizio e regolazione della trascrizione del gene a valle (Juven-Gershon et al., 2008). L’inizio della trascrizione genica è il passo più fondamentale nella regolazione dell’espressione genica. Il nucleo del promotore è un tratto minimo di sequenza di DNA che si connette a TSS e sufficiente per avviare direttamente la trascrizione. La lunghezza del promotore principale varia tipicamente tra 60 e 120 paia di basi (bp).
Il TATA-box è una sottosequenza del promotore che indica ad altre molecole dove inizia la trascrizione. È stata chiamata “TATA-box” poiché la sua sequenza è caratterizzata dalla ripetizione di coppie di basi T e A (TATAAA) (Baker et al., 2003). La stragrande maggioranza degli studi sulla TATA-box sono stati condotti e genomi di Drosophila, tuttavia, elementi simili sono stati trovati in altre specie come archaea e antichi eucarioti (Smale e Kadonaga, 2003). Nel caso umano, il 24% dei geni ha regioni promotore contenenti TATA-box (Yang et al., 2007 Negli eucarioti, il TATA-box si trova a ~ 25 bp a monte del TSS (Xu et al., 2016). È in grado di definire la direzione della trascrizione e indica anche il filamento di DNA da leggere. Proteine chiamate fattori di trascrizione legarsi a diverse regioni non codificanti tra cui TATA-box e reclutare un enzima chiamato RNA polimerasi, che sintetizza l’RNA dal DNA.
A causa dell’importante ruolo dei promotori nella trascrizione genica, la previsione accurata dei siti del promotore diventa un passaggio necessario nell’espressione genica, nell’interpretazione dei modelli e nella costruzione e comprensione la funzionalità delle reti di regolazione genetica. Ci sono stati diversi esperimenti biologici per l’identificazione di promotori come l’analisi mutazionale (Matsumine et al., 1998) e saggi di immunoprecipitazione (Kim et al., 2004; Dahl e Collas, 2008). Tuttavia, questi metodi erano sia costosi che dispendiosi in termini di tempo. Recentemente, con lo sviluppo del sequenziamento di nuova generazione (NGS) (Behjati e Tarpey, 2013) sono stati sequenziati più geni di diversi organismi e i loro elementi genetici sono stati esplorati computazionalmente (Zhang et al., 2011). D’altra parte, l’innovazione della tecnologia NGS ha comportato un drastico calo del costo dell’intero sequenziamento del genoma, quindi sono disponibili più dati di sequenziamento. La disponibilità dei dati attira i ricercatori a sviluppare modelli computazionali per l’attività di predizione del promotore. Tuttavia, è ancora un’attività incompleta e non esiste un software efficiente in grado di prevedere con precisione i promotori.
I predittori dei promotori possono essere classificati in base all’approccio utilizzato in tre gruppi: approccio basato sui segnali, approccio basato sul contenuto e l’approccio basato su GpG. I predittori basati sul segnale si concentrano sugli elementi promotori relativi al sito di legame della RNA polimerasi e ignorano le porzioni non elemento della sequenza. Di conseguenza, l’accuratezza della previsione era debole e non soddisfacente. Esempi di predittori basati sul segnale includono: PromoterScan (Prestridge, 1995) che ha utilizzato le caratteristiche estratte del TATA-box e una matrice pesata di siti di legame del fattore di trascrizione con un discriminatore lineare per classificare le sequenze del promotore da quelle non promotori; Promoter2.0 (Knudsen, 1999) che ha estratto le caratteristiche da diversi box come TATA-Box, CAAT-Box e GC-Box e le ha passate a reti neurali artificiali (ANN) per la classificazione; NNPP2.1 (Reese, 2001) che utilizzava l’elemento iniziatore (Inr) e TATA-Box per l’estrazione delle caratteristiche e una rete neurale a ritardo di tempo per la classificazione, e Down e Hubbard (2002) che utilizzavano TATA-Box e utilizzavano macchine vettoriali di rilevanza (RVM) come classificatore. I predittori basati sul contenuto si basavano sul conteggio della frequenza di k-mer eseguendo una finestra di lunghezza k lungo la sequenza. Tuttavia, questi metodi ignorano le informazioni spaziali delle coppie di basi nelle sequenze. Esempi di predittori basati sul contenuto includono: PromFind (Hutchinson, 1996) che ha utilizzato la frequenza k-mer per eseguire la predizione del promotore dell’esamero; PromoterInspector (Scherf et al., 2000) che ha identificato le regioni contenenti promotori sulla base di un contesto genomico comune dei promotori della polimerasi II mediante scansione per caratteristiche specifiche definite come motivi di lunghezza variabile; MCPromoter1.1 (Ohler et al., 1999) che utilizzava una singola catena di Markov interpolata (IMC) di 5 ° ordine per prevedere le sequenze del promotore.Infine, i predittori basati su GpG hanno utilizzato la posizione delle isole GpG come regione promotrice o la prima regione esone nei geni umani di solito contiene isole GpG (Ioshikhes e Zhang, 2000; Davuluri et al., 2001; Lander et al., 2001; Ponger e Mouchiroud, 2002). Tuttavia, solo il 60% dei promotori contiene isole GpG, quindi l’accuratezza della previsione di questo tipo di predittori non ha mai superato il 60%.
Recentemente sono stati utilizzati approcci basati sulla sequenza per la previsione del promotore. Yang et al. (2017) hanno utilizzato diverse strategie di estrazione delle caratteristiche per acquisire le informazioni sulla sequenza più rilevanti al fine di prevedere le interazioni potenziatore-promotore. Lin et al. (2017) hanno proposto un predittore basato sulla sequenza, denominato “iPro70-PseZNC”, per l’identificazione del promotore sigma70 nel procariota. Allo stesso modo, Bharanikumar et al. (2018) hanno proposto PromoterPredict al fine di prevedere la forza dei promotori di Escherichia coli sulla base di un approccio di regressione multipla dinamica in cui le sequenze erano rappresentate come matrici di peso di posizione (PWM). Kanhere e Bansal (2005) hanno utilizzato le differenze nella stabilità della sequenza del DNA tra le sequenze promotore e non promotore per distinguerle. Xiao et al. (2018) ha introdotto un predittore a due livelli chiamato iPSW (2L) -PseKNC per l’identificazione delle sequenze del promotore nonché la forza dei promotori estraendo le caratteristiche ibride dalle sequenze.
Tutti i predittori sopra menzionati richiedono il dominio- conoscenza al fine di creare a mano le caratteristiche. D’altra parte, gli approcci basati sull’apprendimento profondo consentono di costruire modelli più efficienti utilizzando direttamente dati grezzi (sequenze di DNA / RNA). La rete neurale convoluzionale profonda ha ottenuto risultati all’avanguardia in compiti impegnativi come l’elaborazione di immagini, video, audio e parlato (Krizhevsky et al., 2012; LeCun et al., 2015; Schmidhuber, 2015; Szegedy et al. , 2015). Inoltre, è stato applicato con successo in problemi biologici come DeepBind (Alipanahi et al., 2015), DeepCpG (Angermueller et al., 2017), selezione del punto di diramazione (Nazari et al., 2018), previsione di siti di splicing alternativi (Oubounyt et al., 2018), predizione dei siti di 2 “-Omethylation (Tahir et al., 2018), quantificazione della sequenza del DNA (Quang e Xie, 2016), localizzazione subcellulare della proteina umana (Wei et al., 2018), ecc. La CNN ha recentemente guadagnato un’attenzione significativa nel compito di riconoscimento del promotore. Molto recentemente, Umarov e Solovyev (2017) hanno introdotto CNNprom per la discriminazione delle sequenze del promotore breve, questa architettura basata sulla CNN ha ottenuto risultati elevati nella classificazione delle sequenze promotore e non promotore. Successivamente, questo modello è stato migliorato di Qian et al. (2018) in cui gli autori hanno utilizzato il classificatore SVM (Support Vector Machine) per ispezionare gli elementi di sequenza del promotore più importanti. Successivamente, gli elementi più influenti sono stati mantenuti non compressi mentre si comprimevano quelli meno importanti. Questo processo ha portato a prestazioni migliori. Recentemente, il modello di identificazione del promotore lungo è stato proposto da Umarov et al. (2019) in cui gli autori si sono concentrati sull’identificazione della posizione del TSS.
In tutti i lavori sopra menzionati il set negativo è stato estratto da regioni non promotori del genoma. Sapendo che le sequenze del promotore sono ricche esclusivamente di elementi funzionali specifici come TATA-box che si trova a –30 ~ –25 bp, GC-Box che si trova a –110 ~ –80 bp, CAAT-Box che si trova a – 80 ~ –70 bp, ecc. Ciò si traduce in un’elevata precisione di classificazione a causa dell’enorme disparità tra i campioni positivi e negativi in termini di struttura della sequenza. Inoltre, l’attività di classificazione diventa facile da ottenere, ad esempio, i modelli della CNN si baseranno semplicemente sulla presenza o l’assenza di alcuni motivi nelle loro posizioni specifiche per prendere la decisione sul tipo di sequenza. Pertanto, questi modelli hanno una precisione / sensibilità molto bassa (alto falso positivo) quando vengono testati su sequenze genomiche che hanno motivi promotori ma non sono sequenze promotori. È noto che ci sono più motivi TATAAA nel genoma rispetto a quelli appartenenti alle regioni del promotore. Ad esempio, la sola sequenza di DNA del cromosoma umano 1, ftp://ftp.ensembl.org/pub/release-57/fasta/homo_sapiens/dna/, contiene 151 656 motivi TATAAA. È più del numero massimo approssimato di geni nel genoma umano totale. Come illustrazione di questo problema, notiamo che quando si testano questi modelli su sequenze non promotori che hanno TATA-box, classificano erroneamente la maggior parte di queste sequenze. Pertanto, al fine di generare un classificatore robusto, l’insieme negativo dovrebbe essere selezionato con attenzione in quanto determina le caratteristiche che verranno utilizzate dal classificatore per discriminare le classi. L’importanza di questa idea è stata dimostrata in lavori precedenti come (Wei et al., 2014). In questo lavoro, affrontiamo principalmente questo problema e proponiamo un approccio che integra alcuni dei motivi funzionali di classe positiva nella classe negativa per ridurre la dipendenza del modello da questi motivi.Utilizziamo una CNN combinata con il modello LSTM per analizzare le caratteristiche di sequenza di promotori eucariotici TATA e non TATA umani e murini e costruire modelli computazionali in grado di discriminare accuratamente sequenze di promotori brevi da quelle non promotori.
Materiali e metodi
2.1. Set di dati
I set di dati, utilizzati per l’addestramento e il test del predittore promotore proposto, vengono raccolti da umani e topi. Contengono due classi distintive dei promotori, vale a dire promotori TATA (cioè le sequenze che contengono scatola TATA) e promotori non TATA. Questi set di dati sono stati creati dal database del promotore eucariotico (EPDnew) (Dreos et al., 2012). L’EPDnew è una nuova sezione del noto dataset EPD (Périer et al., 2000) in cui è annotata una raccolta non ridondante di promotori eucariotici POL II in cui il sito di inizio della trascrizione è stato determinato sperimentalmente. Fornisce promotori di alta qualità rispetto alla raccolta di promotori ENSEMBL (Dreos et al., 2012) ed è pubblicamente accessibile all’indirizzo https://epd.epfl.ch//index.php. Abbiamo scaricato le sequenze genomiche del promotore TATA e non TATA per ciascun organismo da EPDnew. Questa operazione ha portato all’ottenimento di quattro set di dati del promotore, vale a dire: Human-TATA, Human-non-TATA, Mouse-TATA e Mouse-non-TATA. Per ciascuno di questi set di dati, viene costruito un set negativo (sequenze non promotori) con la stessa dimensione di quello positivo in base all’approccio proposto come descritto nella sezione seguente. I dettagli sul numero di sequenze del promotore per ciascun organismo sono riportati nella Tabella 1. Tutte le sequenze hanno una lunghezza di 300 bp e sono state estratte da -249 ~ + 50 bp (+1 si riferisce alla posizione TSS). Come controllo di qualità, abbiamo utilizzato 5 volte la convalida incrociata per valutare il modello proposto. In questo caso, 3 volte vengono utilizzate per l’addestramento, 1 piega viene utilizzata per la convalida e la piega rimanente viene utilizzata per il test. Pertanto, il modello proposto viene addestrato 5 volte e viene calcolata la prestazione complessiva del 5 volte.

Tabella 1. Statistiche dei quattro set di dati utilizzati in questo studio.
2.2. Costruzione di set di dati negativi
Per addestrare un modello in grado di eseguire accuratamente la classificazione di sequenze promotore e non promotore, è necessario scegliere attentamente l’insieme negativo (sequenze non promotore). Questo punto è fondamentale per realizzare un modello in grado di generalizzare bene, e quindi in grado di mantenere la sua precisione quando valutato su dataset più impegnativi. Lavori precedenti, come (Qian et al., 2018), costruivano un set negativo selezionando casualmente frammenti da regioni non promotori del genoma. Ovviamente, questo approccio non è del tutto ragionevole perché se non c’è intersezione tra insiemi positivi e negativi. Pertanto, il modello troverà facilmente le caratteristiche di base per separare le due classi. Ad esempio, il motivo TATA può essere trovato in tutte le sequenze positive in una posizione specifica (normalmente 28 bp a monte del TSS, tra –30 e –25 pb nel nostro set di dati). Pertanto, la creazione casuale di un set negativo che non contiene questo motivo produrrà prestazioni elevate in questo set di dati. Tuttavia, il modello non riesce a classificare le sequenze negative che hanno il motivo TATA come promotori. In breve, il principale difetto di questo approccio è che quando si allena un modello di apprendimento profondo si impara solo a discriminare le classi positive e negative in base alla presenza o all’assenza di alcune semplici caratteristiche in posizioni specifiche, il che rende questi modelli impraticabili. In questo lavoro, miriamo a risolvere questo problema stabilendo un metodo alternativo per derivare l’insieme negativo da quello positivo.
Il nostro metodo si basa sul fatto che ogni volta che le caratteristiche sono comuni tra il negativo e il classe positiva il modello tende, quando prende la decisione, a ignorare o ridurre la sua dipendenza da queste caratteristiche (cioè ad assegnare pesi bassi a queste caratteristiche). Invece, il modello è costretto a cercare caratteristiche più profonde e meno evidenti. I modelli di deep learning generalmente soffrono di una lenta convergenza durante l’addestramento su questo tipo di dati. Tuttavia, questo metodo migliora la robustezza del modello e garantisce la generalizzazione. Ricostruiamo l’insieme negativo come segue. Ogni sequenza positiva genera una sequenza negativa. La sequenza positiva è suddivisa in 20 sottosequenze. Quindi, 12 sottosequenze vengono selezionate in modo casuale e sostituite in modo casuale. Le restanti 8 sottosequenze vengono conservate. Questo processo è illustrato nella Figura 1. Applicando questo processo all’insieme positivo si ottengono nuove sequenze non promotore con parti conservate da sequenze promotore (le sottosequenze invariate, 8 sottosequenze su 20). Questi parametri consentono di generare un set negativo che ha il 32 e il 40% delle sue sequenze contenenti porzioni conservate di sequenze promotore. Questo rapporto è risultato essere ottimale per avere un robusto predittore del promotore come spiegato nella sezione 3.2.Poiché le parti conservate occupano le stesse posizioni nelle sequenze negative, i motivi ovvi come TATA-box e TSS sono ora comuni tra i due set con un rapporto del 32 ~ 40%. I loghi di sequenza dei set positivi e negativi per i dati del promotore TATA sia umano che di topo sono mostrati nelle Figure 2 e 3, rispettivamente. Si può vedere che l’insieme positivo e negativo condividono gli stessi motivi di base nelle stesse posizioni come il motivo TATA nella posizione -30 e -25 bp e il TSS nella posizione +1 bp. Pertanto, la formazione è più impegnativa ma il modello risultante si generalizza bene.
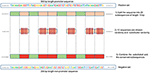
Figura 1. Illustrazione del metodo di costruzione dell’insieme negativo. Il verde rappresenta le sottosequenze conservate casualmente mentre il rosso rappresenta quelle scelte casualmente e sostituite.

Figura 2. Il logo della sequenza nel promotore TATA umano sia per il set positivo (A) che per il set negativo (B). I grafici mostrano la conservazione dei motivi funzionali tra i due insiemi.

Figura 3. Il logo della sequenza nel promotore TATA del mouse sia per il set positivo (A) che per il set negativo (B). I grafici mostrano la conservazione dei motivi funzionali tra i due insiemi.
2.3. I modelli proposti
Proponiamo un modello di apprendimento profondo che combina strati di convoluzione con strati ricorrenti come mostrato nella Figura 4. Accetta una singola sequenza genomica grezza, S = {N1, N2,…, Nl} dove N ∈ {A, C, G, T} el è la lunghezza della sequenza di input, come input e output di un punteggio a valore reale. L’input è codificato a caldo e rappresentato come un vettore unidimensionale con quattro canali. La lunghezza del vettore l = 300 e i quattro canali sono A, C, G e T e rappresentati come (1 0 0 0), (0 1 0 0), (0 0 1 0), (0 0 0 1 ), rispettivamente. Al fine di selezionare il modello più performante, abbiamo utilizzato il metodo di ricerca della griglia per scegliere i migliori iperparametri. Abbiamo provato diverse architetture come solo CNN, solo LSTM, solo BiLSTM, CNN combinato con LSTM. Gli iperparametri sintonizzati sono il numero di livelli di convoluzione, la dimensione del kernel, il numero di filtri in ogni livello, la dimensione del livello di pooling massimo, la probabilità di interruzione e le unità del livello Bi-LSTM.
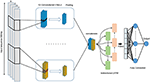
Figura 4. L’architettura del modello DeePromoter proposto.
Il modello proposto inizia con più livelli di convoluzione che sono allineati in parallelo e aiutano ad apprendere i motivi importanti delle sequenze di input con diverse dimensioni della finestra. Usiamo tre strati di convoluzione per promotori non TATA con dimensioni della finestra di 27, 14 e 7 e due strati di convoluzione per promotori TATA con dimensioni di finestra di 27, 14. Tutti gli strati di convoluzione sono seguiti dalla funzione di attivazione ReLU (Glorot et al. , 2011), un livello di pool massimo con una dimensione della finestra di 6 e uno strato di esclusione di probabilità 0,5. Quindi, gli output di questi strati vengono concatenati insieme e inseriti in uno strato di memoria bidirezionale a lungo termine (BiLSTM) (Schuster e Paliwal, 1997) con 32 nodi al fine di catturare le dipendenze tra i motivi appresi dagli strati di convoluzione. Le caratteristiche apprese dopo BiLSTM vengono appiattite e seguite da un abbandono con una probabilità di 0,5. Quindi aggiungiamo due livelli completamente connessi per la classificazione. Il primo ha 128 nodi e seguito da ReLU e dropout con una probabilità di 0,5 mentre il secondo strato viene utilizzato per la previsione con un nodo e la funzione di attivazione del sigmoide. BiLSTM consente alle informazioni di persistere e apprendere le dipendenze a lungo termine di campioni sequenziali come DNA e RNA. Ciò si ottiene tramite la struttura LSTM che è composta da una cella di memoria e tre porte chiamate porte di input, output e dimentica. Queste porte sono responsabili della regolazione delle informazioni nella cella di memoria. Inoltre, l’utilizzo del modulo LSTM aumenta la profondità della rete mentre il numero dei parametri richiesti rimane basso. Avere una rete più profonda consente di estrarre caratteristiche più complesse e questo è l’obiettivo principale dei nostri modelli poiché il set negativo contiene campioni duri.
Il framework Keras viene utilizzato per costruire e addestrare i modelli proposti (Chollet F. et al., 2015). Adam optimizer (Kingma e Ba, 2014) viene utilizzato per aggiornare i parametri con una velocità di apprendimento di 0,001. La dimensione del batch è impostata su 32 e il numero di epoche è impostato su 50. L’arresto anticipato viene applicato in base alla perdita di convalida.
Risultati e discussione
3.1. Misurazioni delle prestazioni
In questo lavoro, utilizziamo le metriche di valutazione ampiamente adottate per valutare le prestazioni dei modelli proposti.Queste metriche sono precisione, richiamo e coefficiente di correlazione Matthew (MCC) e sono definite come segue:
Dove TP è vero positivo e rappresenta sequenze di promotori identificate correttamente, TN è vero negativo e rappresenta sequenze di promotori correttamente rifiutate, FP è falso positivo e rappresenta identificato in modo errato sequenze del promotore e FN è falso negativo e rappresenta sequenze del promotore rifiutate in modo errato.
3.2. Effetto dell’insieme negativo
Analizzando i lavori pubblicati in precedenza per l’identificazione delle sequenze del promotore abbiamo notato che le prestazioni di quei lavori dipendono molto dal modo di preparare l’insieme di dati negativi. Hanno funzionato molto bene sui set di dati che hanno preparato, tuttavia, hanno un alto rapporto di falsi positivi quando valutati su un set di dati più impegnativo che include sequenze non suggeritore con motivi comuni con sequenze promotore. Ad esempio, nel caso del set di dati del promotore TATA, le sequenze generate casualmente non avranno motivo TATA nella posizione -30 e -25 bp, il che a sua volta rende più facile il compito di classificazione. In altre parole, il loro classificatore dipendeva dalla presenza del motivo TATA per identificare la sequenza del promotore e, di conseguenza, è stato facile ottenere prestazioni elevate sui set di dati che hanno preparato. Tuttavia, i loro modelli fallirono drammaticamente quando si trattava di sequenze negative che contenevano motivi TATA (esempi concreti). La precisione è diminuita all’aumentare del tasso di falsi positivi. Semplicemente, hanno classificato queste sequenze come sequenze promotori positive. Un’analisi simile è valida per gli altri motivi promotori. Pertanto, lo scopo principale del nostro lavoro non è solo ottenere prestazioni elevate su un set di dati specifico, ma anche migliorare la capacità del modello di generalizzare bene allenandosi su un set di dati impegnativo.
Per illustrare meglio questo punto, ci alleniamo e testare il nostro modello sui set di dati del promotore TATA umano e di topo con diversi metodi di preparazione dei set negativi. Il primo esperimento viene eseguito utilizzando sequenze negative campionate casualmente da regioni non codificanti del genoma (cioè, simile all’approccio utilizzato nei lavori precedenti). Sorprendentemente, il nostro modello proposto raggiunge un’accuratezza di previsione quasi perfetta (precisione = 99%, richiamo = 99%, Mcc = 98%) e (precisione = 99%, richiamo = 98%, Mcc = 97%) sia per l’uomo che per il topo, rispettivamente . Questi risultati elevati sono previsti, ma la domanda è se questo modello può mantenere le stesse prestazioni se valutato su un set di dati che contiene esempi concreti. La risposta, basata sull’analisi dei modelli precedenti, è no. Il secondo esperimento viene eseguito utilizzando il metodo proposto per la preparazione del set di dati, come spiegato nella sezione 2.2. Prepariamo i set negativi che contengono TATA-box conservati con diverse percentuali come 12, 20, 32 e 40% e l’obiettivo è ridurre il divario tra la precisione e il richiamo. Ciò garantisce che il nostro modello apprenda funzionalità più complesse piuttosto che apprendere solo la presenza o l’assenza di TATA-box. Come mostrato nelle Figure 5A, B, il modello si stabilizza al rapporto 32 ~ 40% per i set di dati del promotore TATA sia umano che di topo.
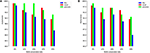
Figura 5. L’effetto di diversi rapporti di conservazione del motivo TATA nel set negativo sulle prestazioni in caso di set di dati del promotore TATA sia per l’uomo (A) che per il topo (B) .
3.3. Risultati e confronto
Negli ultimi anni sono stati proposti molti strumenti di predizione della regione promotrice (Hutchinson, 1996; Scherf et al., 2000; Reese, 2001; Umarov e Solovyev, 2017). Tuttavia, alcuni di questi strumenti non sono pubblicamente disponibili per i test e alcuni di essi richiedono ulteriori informazioni oltre alle sequenze genomiche grezze. In questo studio, confrontiamo le prestazioni dei nostri modelli proposti con l’attuale lavoro all’avanguardia, CNNProm, che è stato proposto da Umarov e Solovyev (2017) come mostrato nella Tabella 2. In generale, i modelli proposti, DeePromoter, chiaramente sovraperforma CNNProm in tutti i set di dati con tutte le metriche di valutazione. Più specificamente, DeePromoter migliora la precisione, il richiamo e l’MCC nel caso del set di dati TATA umano di 0,18, 0,04 e 0,26, rispettivamente. Nel caso di set di dati umani non TATA, DeePromoter migliora la precisione di 0,39, il richiamo di 0,12 e MCC di 0,66. Allo stesso modo, DeePromoter migliora la precisione e MCC nel caso del set di dati TATA del mouse di 0,24 e 0,31, rispettivamente. Nel caso del set di dati del mouse non TATA, DeePromoter migliora la precisione di 0,37, il richiamo di 0,04 e MCC di 0,65. Questi risultati confermano che CNNProm non riesce a rifiutare sequenze negative con il promotore TATA, quindi ha un alto numero di falsi positivi. D’altra parte, i nostri modelli sono in grado di affrontare questi casi con maggiore successo e il tasso di falsi positivi è inferiore rispetto a CNNProm.

Tabella 2. Confronto del DeePromoter con lo stato di -metodo dell’arte.
Per ulteriori analisi, studiamo l’effetto dell’alternanza dei nucleotidi in ciascuna posizione sul punteggio di output. Ci concentriamo sulla regione –40 e 10 bp poiché ospita la parte più importante della sequenza del promotore. Per ciascuna sequenza del promotore nel set di test, eseguiamo la scansione computazionale delle mutazioni per valutare l’effetto della mutazione di ogni base della sottosequenza di input (150 sostituzioni nell’intervallo –40 ~ 10 bp sottosequenza). Ciò è illustrato nelle Figure 6 e 7 rispettivamente per i set di dati TATA umani e di topo. Il colore blu rappresenta un calo del punteggio di output dovuto alla mutazione mentre il colore rosso rappresenta l’incremento del punteggio dovuto alla mutazione. Notiamo che l’alterazione dei nucleotidi in C o G nella regione –30 e –25 bp riduce significativamente il punteggio di output. Questa regione è TATA-box che è un motivo funzionale molto importante nella sequenza del promotore. Pertanto, il nostro modello è in grado di trovare con successo l’importanza di questa regione. Nel resto delle posizioni, i nucleotidi C e G sono più preferibili di A e T, specialmente nel caso del topo. Ciò può essere spiegato dal fatto che la regione del promotore ha più nucleotidi C e G di A e T (Shi e Zhou, 2006).

Figura 6. La mappa di salienza della regione da –40 bp a 10 bp, che include il TATA-box, in caso di sequenze di promotori TATA umani.

Figura 7. La mappa di salienza della regione da –40 bp a 10 bp, che include il TATA-box, in caso di sequenze di promotori TATA di topo.
Conclusione
Una previsione accurata delle sequenze del promotore è essenziale per comprendere il meccanismo alla base del processo di regolazione genica. In questo lavoro, abbiamo sviluppato DeePromoter, che si basa su una combinazione di rete neurale di convoluzione e LSTM bidirezionale, per prevedere le brevi sequenze del promotore di eucarioti in caso di uomo e topo sia per promotore TATA che non TATA. La componente essenziale di questo lavoro è stata quella di superare il problema della bassa precisione (alto tasso di falsi positivi) notato negli strumenti sviluppati in precedenza a causa della dipendenza da alcune caratteristiche / motivi evidenti nella sequenza quando si classificano sequenze promotore e non promotore. In questo lavoro, eravamo particolarmente interessati alla costruzione di un set negativo rigido che spinga i modelli verso l’esplorazione della sequenza per caratteristiche profonde e rilevanti invece di distinguere solo le sequenze promotore e non promotore in base all’esistenza di alcuni motivi funzionali. I principali vantaggi dell’utilizzo di DeePromoter è che riduce significativamente il numero di previsioni false positive, ottenendo un’elevata precisione su set di dati impegnativi. DeePromoter ha sovraperformato il metodo precedente non solo nelle prestazioni ma anche nel superare il problema delle previsioni di falsi positivi elevati. Si prevede che questo framework possa essere utile nelle applicazioni legate alla droga e nel mondo accademico.
Contributi dell’autore
MO e ZL hanno preparato il set di dati, concepito l’algoritmo, condotto l’esperimento e analisi. MO e HT hanno preparato il server web e hanno scritto il manoscritto con il supporto di ZL e KC. Tutti gli autori hanno discusso i risultati e hanno contribuito al manoscritto finale.
Finanziamento
Questa ricerca è stata sostenuta dal Brain Research Program della National Research Foundation (NRF) finanziato dal governo coreano ( MSIT) (n. NRF-2017M3C7A1044815).
Conflitto di interessi
Gli autori dichiarano che la ricerca è stata condotta in assenza di rapporti commerciali o finanziari che potrebbero essere interpretati come un potenziale conflitto di interessi.
Bharanikumar, R., Premkumar, KAR e Palaniappan, A. (2018). Promotore predittivo: la modellazione basata sulla sequenza della forza del promotore di escherichia coli σ70 produce una dipendenza logaritmica tra la forza del promotore e la sequenza. PeerJ 6: e5862. doi: 10.7717 / peerj.5862
Estratto di PubMed | Testo completo CrossRef | Google Scholar
Glorot, X., Bordes, A. e Bengio, Y. (2011). “Deep sparse rectifier neural networks”, in Atti della quattordicesima conferenza internazionale sull’intelligenza artificiale e le statistiche, (Fort Lauderdale, FL:) 315–323.
Google Scholar
Hutchinson, G. (1996). La previsione delle regioni del promotore dei vertebrati utilizzando l’analisi della frequenza differenziale degli esameri. Bioinformatica 12, 391–398.
Estratto di PubMed | Google Scholar
Kingma, DP e Ba, J. (2014). Adam: a method for stochastic optimization. arXiv preprint arXiv: 1412.6980.
Google Scholar
Knudsen, S. (1999). Promotore2. 0: per il riconoscimento di sequenze di promotori di polii. Bioinformatica 15, 356-361.
Estratto di PubMed | Google Scholar
Ponger, L. e Mouchiroud, D. (2002). Cpgprod: identificazione di isole cpg associate a siti di inizio trascrizione in grandi sequenze di mammiferi genomici. Bioinformatica 18, 631–633. doi: 10.1093 / bioinformatics / 18.4.631
Estratto di PubMed | Testo completo CrossRef | Google Scholar
Quang, D. and Xie, X. (2016). Danq: una rete neurale profonda ibrida convoluzionale e ricorrente per quantificare la funzione delle sequenze di DNA. Ris. Acidi nucleici 44, e107 – e107. doi: 10.1093 / nar / gkw226
Estratto di PubMed | Testo completo CrossRef | Google Scholar
Umarov, R. K. e Solovyev, V. V. (2017). Riconoscimento di promotori procariotici ed eucariotici mediante reti neurali convoluzionali di apprendimento profondo. PLoS ONE 12: e0171410. doi: 10.1371 / journal.pone.0171410
Estratto di PubMed | Testo completo CrossRef | Google Scholar