遺伝学の最前線
はじめに
プロモーターは、ゲノムの非コード領域に属する重要な要素です。それらは主に遺伝子の活性化または抑制を制御します。それらは、遺伝子の転写開始部位(TSS)の近くおよび上流に位置します。遺伝子のプロモーター隣接領域には、提供するタンパク質の認識部位として機能する多くの重要な短いDNA要素およびモチーフ(5および15塩基長)が含まれる場合があります。下流遺伝子の転写の適切な開始と調節(Juven-Gershon et al。、2008)。遺伝子転写の開始は、遺伝子発現の調節における最も基本的なステップです。プロモーターコアは、TSSを結合する最小限のDNA配列であり、転写を直接開始するのに十分です。コアプロモーターの長さは通常、60〜120塩基対(bp)の範囲です。
TATAボックスは、転写が始まる場所を他の分子に示すプロモーターサブシーケンスです。その配列がTおよびA塩基対(TATAAA)を繰り返すことを特徴とするため、「TATAボックス」と名付けられました(Baker et al。、2003)。TATAボックスに関する研究の大部分は、ヒト、酵母、ただし、ショウジョウバエのゲノムは、古細菌や古代の真核生物などの他の種でも同様の要素が見つかっています(Smale and Kadonaga、2003)。ヒトの場合、遺伝子の24%にTATAボックスを含むプロモーター領域があります(Yang et al。、2007 )真核生物では、TATAボックスはTSSの約25 bp上流にあります(Xu et al。、2016)。転写の方向を定義することができ、読み取るDNA鎖も示します。転写因子と呼ばれるタンパク質TATAボックスを含むいくつかの非コード領域に結合し、DNAからRNAを合成するRNAポリメラーゼと呼ばれる酵素を動員します。
遺伝子転写におけるプロモーターの重要な役割により、プロモーター部位の正確な予測は次のようになります。遺伝子発現、パターン解釈、構築と理解に必要なステップ遺伝子調節ネットワークの機能を利用する。突然変異分析(Matsumine et al。、1998)や免疫沈降アッセイ(Kim et al。、2004; Dahl and Collas、2008)など、プロモーターを同定するためのさまざまな生物学的実験がありました。ただし、これらの方法は費用と時間がかかりました。最近、次世代シーケンシング(NGS)の開発に伴い(Behjati and Tarpey、2013)、さまざまな生物のより多くの遺伝子がシーケンシングされ、それらの遺伝子要素がコンピューターで探索されました(Zhang et al。、2011)。一方、NGSテクノロジーの革新により、全ゲノムシーケンスのコストが劇的に低下したため、より多くのシーケンスデータを利用できます。データの可用性は、プロモーター予測タスクの計算モデルを開発するために研究者を引き付けます。ただし、それはまだ不完全なタスクであり、プロモーターを正確に予測できる効率的なソフトウェアはありません。
プロモーター予測子は、利用されたアプローチに基づいて、信号ベースのアプローチ、コンテンツベースのアプローチの3つのグループに分類できます。 、およびGpGベースのアプローチ。シグナルベースの予測因子は、RNAポリメラーゼ結合部位に関連するプロモーター要素に焦点を当て、配列の非要素部分を無視します。その結果、予測精度が弱く、満足のいくものではありませんでした。シグナルベースの予測因子の例は次のとおりです。TATAボックスの抽出された特徴と線形弁別器を備えた転写因子結合部位の加重行列を使用してプロモーター配列を非プロモーター配列から分類するPromoterScan(Prestridge、1995)。 Promoter2.0(Knudsen、1999)は、TATA-Box、CAAT-Box、GC-Boxなどのさまざまなボックスから特徴を抽出し、分類のために人工ニューラルネットワーク(ANN)に渡しました。特徴抽出にイニシエーター要素(Inr)とTATA-Boxを使用し、分類に時間遅延ニューラルネットワークを使用したNNPP2.1(Reese、2001)、およびTATA-Boxを使用して関連性ベクトルマシンを使用したDown and Hubbard(2002) (RVM)分類器として。コンテンツベースの予測子は、シーケンス全体でk長のウィンドウを実行することによってk-merの頻度をカウントすることに依存していました。ただし、これらのメソッドは、シーケンス内の塩基対の空間情報を無視します。コンテンツベースの予測子の例は次のとおりです。PromFind(Hutchinson、1996)は、k-mer頻度を使用してヘキサマープロモーター予測を実行しました。可変長モチーフとして定義された特定の特徴をスキャンすることにより、ポリメラーゼIIプロモーターの一般的なゲノムコンテキストに基づいてプロモーターを含む領域を特定したPromoterInspector(Scherf et al。、2000)。 MCPromoter1.1(Ohler et al。、1999)は、プロモーター配列を予測するために5次の単一の補間マルコフ連鎖(IMC)を使用しました。最後に、GpGベースの予測子は、プロモーター領域またはヒト遺伝子の最初のエクソン領域としてGpGアイランドの位置を利用し、通常はGpGアイランドを含みます(Ioshikhes and Zhang、2000; Davuluri et al。、2001; Lander et al。、2001; Ponger and Mouchiroud、2002)。ただし、プロモーターの60%のみがGpGアイランドを含んでいるため、この種の予測子の予測精度は60%を超えることはありません。
最近、プロモーターの予測にシーケンスベースのアプローチが利用されています。ヤンら。 (2017)エンハンサーとプロモーターの相互作用を予測するために、さまざまな特徴抽出戦略を利用して、最も関連性の高い配列情報を取得しました。リンら。 (2017)原核生物におけるsigma70プロモーターの同定のために、「iPro70-PseZNC」と名付けられた配列ベースの予測因子を提案しました。同様に、Bharanikumar etal。 (2018)シーケンスが位置重み行列(PWM)として表される動的重回帰アプローチに基づいて、大腸菌プロモーターの強度を予測するためにPromoterPredictを提案しました。 Kanhere and Bansal(2005)は、プロモーター配列と非プロモーター配列のDNA配列の安定性の違いを利用して、それらを区別しました。 Xiao etal。 (2018)iPSW(2L)と呼ばれる2層の予測子を導入しました-プロモーター配列の識別と配列からハイブリッド機能を抽出することによるプロモーターの強度のためのPseKNC。
前述の予測子はすべてドメインを必要とします-機能を手作りするための知識。一方、ディープラーニングベースのアプローチでは、生データ(DNA / RNAシーケンス)を直接使用して、より効率的なモデルを構築できます。深い畳み込みニューラルネットワークは、画像、ビデオ、オーディオ、音声の処理などの困難なタスクで最先端の結果を達成しました(Krizhevsky et al。、2012; LeCun et al。、2015; Schmidhuber、2015; Szegedy etal。 、2015)。さらに、DeepBind(Alipanahi et al。、2015)、DeepCpG(Angermueller et al。、2017)、分岐点選択(Nazari et al。、2018)、選択的スプライシング部位予測(Oubounyt)などの生物学的問題にうまく適用されました。 et al。、2018)、2 “-O-メチル化部位予測(Tahir et al。、2018)、DNA配列定量化(Quang and Xie、2016)、ヒトタンパク質細胞内局在(Wei et al。、2018)など。 CNNは最近、プロモーター認識タスクで大きな注目を集めました。ごく最近、Umarov and Solovyev(2017)は短いプロモーター配列の識別にCNNpromを導入し、このCNNベースのアーキテクチャは、プロモーター配列と非プロモーター配列の分類で高い結果を達成しました。その後、このモデルは改善されました。 Qian et al。(2018)によると、著者はサポートベクターマシン(SVM)分類子を使用して、最も重要なプロモーター配列要素を検査しました。次に、最も影響力のある要素を非圧縮に保ち、重要度の低い要素を圧縮しました。このプロセスにより、パフォーマンスが向上しました。最近、長いプロモーター同定モデルがUmarovらによって提案されました。 (2019)著者はTSS位置の特定に焦点を合わせました。
上記のすべての研究において、ネガティブセットはゲノムの非プロモーター領域から抽出されました。プロモーター配列は、–30〜–25 bpにあるTATA-box、–110〜–80 bpにあるGC-Box、–にあるCAAT-Boxなどの特定の機能要素のみが豊富であることを知っています。 80〜–70 bpなど。これにより、シーケンス構造の点でポジティブサンプルとネガティブサンプルの間に大きな差異があるため、高い分類精度が得られます。さらに、分類タスクを簡単に実行できます。たとえば、CNNモデルは、特定の位置にあるモチーフの有無に依存して、シーケンスタイプを決定します。したがって、これらのモデルは、プロモーターモチーフを持っているがプロモーター配列ではないゲノム配列でテストした場合、精度/感度が非常に低くなります(偽陽性が高くなります)。ゲノムにはプロモーター領域に属するものよりも多くのTATAAAモチーフがあることはよく知られています。たとえば、ヒト1番染色体のDNA配列、ftp://ftp.ensembl.org/pub/release-57/fasta/homo_sapiens/dna/だけでも、151,656個のTATAAAモチーフが含まれています。これは、ヒトゲノム全体における遺伝子の概算最大数を超えています。この問題の例として、TATAボックスを持つ非プロモーターシーケンスでこれらのモデルをテストすると、これらのシーケンスのほとんどが誤って分類されることがわかります。したがって、堅牢な分類器を生成するには、クラスを区別するために分類器によって使用される特徴を決定するため、ネガティブセットを慎重に選択する必要があります。このアイデアの重要性は、(Wei et al。、2014)などの以前の作品で実証されています。この作業では、主にこの問題に対処し、ポジティブクラスの機能モチーフのいくつかをネガティブクラスに統合して、モデルのこれらのモチーフへの依存を減らすアプローチを提案します。CNNとLSTMモデルを組み合わせて、ヒトとマウスのTATAおよび非TATA真核生物プロモーターの配列特性を分析し、短いプロモーター配列と非プロモーター配列を正確に区別できる計算モデルを構築します。
材料と方法
2.1。データセット
提案されたプロモーター予測子のトレーニングとテストに使用されるデータセットは、人間とマウスから収集されます。それらは、プロモーターの2つの異なるクラス、すなわちTATAプロモーター(すなわち、TATAボックスを含む配列)と非TATAプロモーターを含みます。これらのデータセットは、真核生物プロモーターデータベース(EPDnew)から作成されました(Dreos et al。、2012)。 EPDnewは、よく知られているEPDデータセット(Périeretal。、2000)の下にある新しいセクションで、転写開始部位が実験的に決定された真核生物のPOLIIプロモーターの非冗長コレクションに注釈が付けられています。 ENSEMBLプロモーターコレクション(Dreos et al。、2012)と比較して高品質のプロモーターを提供し、https://epd.epfl.ch//index.phpで公開されています。 EPDnewから各生物のTATAおよび非TATAプロモーターゲノム配列をダウンロードしました。この操作により、Human-TATA、Human-non-TATA、Mouse-TATA、およびMouse-non-TATAの4つのプロモーターデータセットが取得されました。これらのデータセットのそれぞれについて、次のセクションで説明するように、提案されたアプローチに基づいて、ポジティブセットと同じサイズのネガティブセット(非プロモーターシーケンス)が構築されます。各生物のプロモーター配列数の詳細を表1に示します。すべての配列の長さは300bpで、-249〜 + 50 bpから抽出されました(+1はTSS位置を示します)。品質管理として、提案されたモデルを評価するために5分割交差検定を使用しました。この場合、3倍はトレーニングに使用され、1倍は検証に使用され、残りの折りはテストに使用されます。したがって、提案されたモデルは5回トレーニングされ、5倍の全体的なパフォーマンスが計算されます。

表1.この調査で使用した4つのデータセットの統計。
2.2。ネガティブデータセットの構築
プロモーターと非プロモーター配列の分類を正確に実行できるモデルをトレーニングするには、ネガティブセット(非プロモーター配列)を慎重に選択する必要があります。この点は、モデルを適切に一般化できるようにするために重要であり、したがって、より困難なデータセットで評価したときにその精度を維持することができます。 (Qian et al。、2018)などの以前の研究では、ゲノムの非プロモーター領域からフラグメントをランダムに選択することにより、ネガティブセットを構築しました。明らかに、正と負の集合の間に共通部分がない場合、このアプローチは完全に合理的ではありません。したがって、モデルは2つのクラスを分離するための基本的な機能を簡単に見つけることができます。たとえば、TATAモチーフは、特定の位置(通常、TSSの上流28 bp、データセットでは–30〜–25 pb)のすべての陽性配列に見られます。したがって、このモチーフを含まないネガティブセットをランダムに作成すると、このデータセットで高いパフォーマンスが得られます。ただし、このモデルでは、TATAモチーフを持つ負の配列をプロモーターとして分類できません。簡単に言うと、このアプローチの主な欠点は、深層学習モデルをトレーニングするときに、特定の位置でのいくつかの単純な機能の有無に基づいてポジティブクラスとネガティブクラスを区別することだけを学習するため、これらのモデルが実行不可能になることです。この作業では、ポジティブセットからネガティブセットを導出する代替方法を確立することにより、この問題を解決することを目指しています。
私たちの方法は、ネガティブとネガティブの間で特徴が共通であるという事実に基づいています。ポジティブクラスは、決定を行うときに、モデルがこれらの機能への依存を無視または減らす(つまり、これらの機能に低い重みを割り当てる)傾向があります。代わりに、モデルはより深く、あまり目立たない機能を検索することを余儀なくされます。深層学習モデルは、通常、このタイプのデータのトレーニング中に収束が遅くなります。ただし、この方法ではモデルの堅牢性が向上し、一般化が保証されます。ネガティブセットを次のように再構築します。各正のシーケンスは、1つの負のシーケンスを生成します。正のシーケンスは20のサブシーケンスに分割されます。次に、12個のサブシーケンスがランダムに選択され、ランダムに置換されます。残りの8つのサブシーケンスは保存されます。このプロセスを図1に示します。このプロセスをポジティブセットに適用すると、プロモーター配列からの保存された部分を持つ新しい非プロモーター配列が得られます(変更されていないサブシーケンス、20のうち8つのサブシーケンス)。これらのパラメーターにより、プロモーター配列の保存部分を含む配列の32%と40%を持つネガティブセットを生成できます。この比率は、セクション3.2で説明されているように、堅牢なプロモーター予測子を持つのに最適であることがわかります。保存された部分がネガティブシーケンスで同じ位置を占めるため、TATAボックスやTSSなどの明らかなモチーフが2つのセット間で32〜40%の比率で共通になりました。ヒトとマウスの両方のTATAプロモーターデータのポジティブセットとネガティブセットのシーケンスロゴをそれぞれ図2、3に示します。ポジティブセットとネガティブセットは、-30および–25 bpの位置のTATAモチーフ、+ 1 bpの位置のTSSなど、同じ位置で同じ基本モチーフを共有していることがわかります。したがって、トレーニングはより困難ですが、結果のモデルは一般化されます。
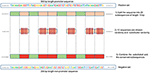
図1.ネガティブセットの構築方法の図。緑はランダムに保存されたサブシーケンスを表し、赤はランダムに選択および置換されたサブシーケンスを表します。

図2.ポジティブセット(A)とネガティブセット(B)の両方のヒトTATAプロモーターのシーケンスロゴ。プロットは、2つのセット間の機能モチーフの保存を示しています。

図3.ポジティブセット(A)とネガティブセット(B)の両方のマウスTATAプロモーターのシーケンスロゴ。プロットは、2つのセット間の機能モチーフの保存を示しています。
2.3。提案されたモデル
図4に示すように、畳み込み層と反復層を組み合わせた深層学習モデルを提案します。これは、単一の生のゲノム配列S = {N1、N2、…、Nl}を受け入れます。ここでN ∈{A、C、G、T}であり、lは入力としての入力シーケンスの長さであり、実数値のスコアを出力します。入力はワンホットエンコードされ、4つのチャネルを持つ1次元ベクトルとして表されます。ベクトルの長さl = 300および4つのチャネルはA、C、G、およびTであり、(1 0 0 0)、(0 1 0 0)、(0 0 1 0)、(0 0 0 1 )、それぞれ。最高のパフォーマンスを発揮するモデルを選択するために、グリッド検索方法を使用して最高のハイパーパラメータを選択しました。 CNNのみ、LSTMのみ、BiLSTMのみ、CNNとLSTMの組み合わせなど、さまざまなアーキテクチャを試しました。調整されたハイパーパラメーターは、畳み込みレイヤーの数、カーネルサイズ、各レイヤーのフィルターの数、最大プーリングレイヤーのサイズ、ドロップアウト確率、およびBi-LSTMレイヤーの単位です。
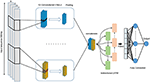
図4.提案されたDeePromoterモデルのアーキテクチャ。
提案されたモデルは、並列に配置された複数の畳み込みレイヤーから始まり、さまざまなウィンドウサイズの入力シーケンスの重要なモチーフの学習に役立ちます。ウィンドウサイズが27、14、および7の非TATAプロモーターには3つの畳み込み層を使用し、ウィンドウサイズが27、14のTATAプロモーターには2つの畳み込み層を使用します。すべての畳み込み層の後にReLU活性化機能が続きます(Glorot etal。 、2011)、ウィンドウサイズが6の最大プーリング層、および確率0.5のドロップアウト層。次に、これらのレイヤーの出力が連結され、畳み込みレイヤーから学習されたモチーフ間の依存関係をキャプチャするために、32ノードの双方向長短期記憶(BiLSTM)(Schuster and Paliwal、1997)レイヤーに送られます。 BiLSTMの後に学習された機能は平坦化され、0.5の確率でドロップアウトが続きます。次に、分類のために2つの完全に接続されたレイヤーを追加します。最初の層には128個のノードがあり、その後に確率0.5のReLUとドロップアウトが続き、2番目の層は1つのノードとシグモイド活性化関数による予測に使用されます。 BiLSTMを使用すると、情報を保持し、DNAやRNAなどの連続サンプルの長期的な依存関係を学習できます。これは、メモリセルと、入力、出力、および忘却ゲートと呼ばれる3つのゲートで構成されるLSTM構造によって実現されます。これらのゲートは、メモリセル内の情報を調整する役割を果たします。さらに、LSTMモジュールを利用すると、必要なパラメータの数を少なくしながら、ネットワークの深さが増します。より深いネットワークを持つことで、より複雑な特徴を抽出できます。ネガティブセットにはハードサンプルが含まれているため、これがモデルの主な目的です。
Kerasフレームワークは、提案されたモデルの構築とトレーニングに使用されます(Chollet F. et al。、2015)。 Adamオプティマイザー(Kingma and Ba、2014)は、0.001の学習率でパラメーターを更新するために使用されます。バッチサイズは32に設定され、エポック数は50に設定されます。早期停止は検証損失に基づいて適用されます。
結果と考察
3.1。パフォーマンス測定
この作業では、提案されたモデルのパフォーマンスを評価するために、広く採用されている評価指標を使用します。これらの指標は適合率、再現率、マシュー相関係数(MCC)であり、次のように定義されます。
TPが真陽性で正しく識別されたプロモーター配列を表す場合、TNが真陰性で正しく拒否されたプロモーター配列を表す場合、FPが偽陽性であり、誤って識別されたことを表すプロモーター配列であり、FNは偽陰性であり、誤って拒否されたプロモーター配列を表します。
3.2。ネガティブセットの効果
プロモーター配列の同定のために以前に公開された作品を分析すると、これらの作品のパフォーマンスはネガティブデータセットの準備方法に大きく依存することがわかりました。彼らは準備したデータセットで非常にうまく機能しましたが、プロモーター配列と共通のモチーフを持つ非プロンプト配列を含むより挑戦的なデータセットで評価した場合、高い偽陽性率を持っています。たとえば、TATAプロモーターデータセットの場合、ランダムに生成された配列には、-30および–25 bpの位置にTATAモチーフがないため、分類のタスクが容易になります。言い換えれば、彼らの分類子はプロモーター配列を識別するためにTATAモチーフの存在に依存しており、その結果、彼らが準備したデータセットで高いパフォーマンスを達成するのは簡単でした。しかし、TATAモチーフを含むネガティブシーケンスを処理する場合、それらのモデルは劇的に失敗しました(難しい例)。偽陽性率が増加すると、精度が低下しました。簡単に言えば、彼らはこれらの配列を陽性プロモーター配列として分類しました。同様の分析は、他のプロモーターモチーフにも当てはまります。したがって、私たちの仕事の主な目的は、特定のデータセットで高いパフォーマンスを達成するだけでなく、挑戦的なデータセットでトレーニングすることにより、一般化のモデル能力を強化することです。
この点をさらに説明するために、トレーニングとネガティブセットの準備のさまざまな方法を使用して、ヒトとマウスのTATAプロモーターデータセットでモデルをテストします。最初の実験は、ゲノムの非コード領域からランダムにサンプリングされたネガティブシーケンスを使用して実行されます(つまり、以前の作業で使用されたアプローチと同様)。驚くべきことに、提案されたモデルは、人間とマウスの両方で、ほぼ完全な予測精度(精度= 99%、再現率= 99%、Mcc = 98%)と(精度= 99%、再現率= 98%、Mcc = 97%)をそれぞれ達成します。 。これらの高い結果が期待されますが、問題は、難しい例があるデータセットで評価したときに、このモデルが同じパフォーマンスを維持できるかどうかです。以前のモデルの分析に基づく答えはノーです。 2番目の実験は、セクション2.2で説明したように、データセットを準備するための提案された方法を使用して実行されます。 12、20、32、40%などのさまざまなパーセンテージで保存されたTATAボックスを含むネガティブセットを準備します。目標は、精度とリコールの間のギャップを減らすことです。これにより、TATAボックスの有無だけを学習するのではなく、モデルがより複雑な機能を学習するようになります。図5A、Bに示すように、モデルは、ヒトとマウスの両方のTATAプロモーターデータセットで32〜40%の比率で安定します。
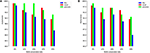
図5.ヒト(A)とマウス(B)の両方のTATAプロモーターデータセットの場合のパフォーマンスに対するネガティブセットのTATAモチーフの異なる保存率の影響。
3.3。結果と比較
過去数年間、多くのプロモーター領域予測ツールが提案されてきました(Hutchinson、1996; Scherf et al。、2000; Reese、2001; Umarov and Solovyev、2017)。ただし、これらのツールの中には、テスト用に公開されていないものもあり、生のゲノム配列以外にさらに多くの情報が必要なものもあります。この研究では、提案されたモデルのパフォーマンスを、表2に示すようにUmarov and Solovyev(2017)によって提案された現在の最先端の作業CNNPromと比較します。一般に、提案されたモデル、DeePromoter、すべての評価指標を備えたすべてのデータセットで、CNNPromを明らかに上回っています。より具体的には、DeePromoterは、人間のTATAデータセットの場合の適合率、再現率、およびMCCをそれぞれ0.18、0.04、および0.26改善します。人間の非TATAデータセットの場合、DeePromoterは精度を0.39、リコールを0.12、MCCを0.66向上させます。同様に、DeePromoterは精度を向上させ、マウスTATAデータセットの場合のMCCはそれぞれ0.24と0.31向上します。マウスの非TATAデータセットの場合、DeePromoterは精度を0.37、リコールを0.04、MCCを0.65向上させます。これらの結果は、CNNPromがTATAプロモーターで陰性配列を拒否できないことを確認しているため、偽陽性が高くなっています。一方、私たちのモデルはこれらのケースをよりうまく処理することができ、偽陽性率はCNNPromと比較して低くなっています。

表2.DeePromoterと最新の状態の比較-最先端の方法。
さらに分析するために、出力スコアに対する各位置のヌクレオチドの交互化の影響を調べます。プロモーター配列の最も重要な部分をホストする領域–40および10bpに焦点を当てます。テストセット内の各プロモーター配列について、計算による突然変異スキャンを実行して、入力サブシーケンスのすべての塩基を突然変異させる効果を評価します(–40〜10 bpサブシーケンスの間隔で150回の置換)。これは、ヒトとマウスのTATAデータセットについてそれぞれ図6、7に示されています。青い色は突然変異による出力スコアの低下を表し、赤い色は突然変異によるスコアの増分を表します。 –30および–25 bpの領域でヌクレオチドをCまたはGに変更すると、出力スコアが大幅に低下することがわかります。この領域は、プロモーター配列の非常に重要な機能モチーフであるTATAボックスです。したがって、私たちのモデルは、この領域の重要性をうまく見つけることができます。残りの位置では、特にマウスの場合、CおよびGヌクレオチドがAおよびTよりも好ましい。これは、プロモーター領域がAおよびTよりも多くのCおよびGヌクレオチドを持っているという事実によって説明できます(Shi and Zhou、2006)。

図6.ヒトTATAプロモーター配列の場合、TATAボックスを含む–40bpから10bpの領域の顕著性マップ。

図7。マウスTATAプロモーター配列の場合のTATAボックスを含む–40bpから10bpの領域の顕著性マップ。
結論
プロモーター配列の正確な予測は、遺伝子調節プロセスの根底にあるメカニズムを理解するために不可欠です。この作業では、畳み込みニューラルネットワークと双方向LSTMの組み合わせに基づくDeePromoterを開発し、TATAプロモーターと非TATAプロモーターの両方についてヒトとマウスの場合の短い真核生物プロモーター配列を予測しました。この作業の重要な要素は、プロモーター配列と非プロモーター配列を分類する際に、配列内のいくつかの明らかな機能/モチーフに依存しているため、以前に開発されたツールで気付いた低精度(高い偽陽性率)の問題を克服することでした。この作業では、いくつかの機能モチーフの存在に基づいてプロモーター配列と非プロモーター配列を区別するだけでなく、深く関連する特徴の配列を探索する方向にモデルを駆動するハードネガティブセットを構築することに特に興味がありました。 DeePromoterを使用する主な利点は、困難なデータセットで高精度を達成しながら、誤検出予測の数を大幅に削減できることです。 DeePromoterは、パフォーマンスだけでなく、高い誤検知予測の問題を克服する点でも、以前の方法を上回りました。このフレームワークは、薬物関連のアプリケーションや学界で役立つ可能性があると予測されています。
著者の貢献
MOとZLは、データセットを準備し、アルゴリズムを考案し、実験を実行し、分析。 MOとHTはウェブサーバーを準備し、ZLとKCの支援を受けて原稿を書きました。すべての著者が結果について話し合い、最終原稿に貢献しました。
資金提供
この研究は、韓国政府が資金提供した国立研究財団(NRF)の脳研究プログラムによって支援されました( MSIT)(No。NRF-2017M3C7A1044815)。
利害の衝突に関する声明
著者は、研究は、次のように解釈される可能性のある商業的または金銭的関係がない状態で行われたと宣言します。潜在的な利益相反。
Bharanikumar、R.、Premkumar、KAR、およびPalaniappan、A。(2018)。プロモーター予測:大腸菌σ70プロモーター強度の配列ベースのモデリングにより、プロモーター強度と配列の間に対数依存性が生じます。 PeerJ 6:e5862。 doi:10.7717 / peerj.5862
PubMed要約| CrossRef全文| Google Scholar
Glorot、X.、Bordes、A。、およびBengio、Y。(2011)。 「人工知能と統計に関する第14回国際会議の議事録」(フロリダ州フォートローダーデール:)315–323の「深く疎な正規化線形ネットワーク」。
Google Scholar
Hutchinson、G。(1996)。微分六量体周波数分析を使用した脊椎動物プロモーター領域の予測。Bioinformatics12、391–398。
PubMed Abstract | Google Scholar
Kingma、DP and Ba、J。(2014)。Adam:確率的最適化の方法。arXivpreprintarXiv:1412.6980。
Google Scholar
Knudsen、S。(1999)。Promoter2.0:poliiプロモーター配列の認識用。Bioinformatics15、356–361。
PubMedの要約| Google Scholar
Ponger、L。and Mouchiroud、D。(2002) Cpgprod:大きなゲノム哺乳類配列の転写開始部位に関連するcpgアイランドを特定します。 バイオインフォマティクス18、631〜633。 doi:10.1093 / bioinformatics / 18.4.631
PubMed要約| CrossRef全文| Google Scholar
Quang、D。and Xie、X。(2016) Danq:dnaシーケンスの機能を定量化するためのハイブリッド畳み込みおよび反復ディープニューラルネットワーク。 核酸解像度。 44、e107〜e107。 doi:10.1093 / nar / gkw226
PubMed要約| CrossRef全文| Google Scholar
Umarov、R。K. and Solovyev、V。V.(2017) 畳み込み深層学習ニューラルネットワークを使用した原核生物および真核生物のプロモーターの認識。 PLoS ONE 12:e0171410。 doi:10.1371 / journal.pone.0171410
PubMed要約| CrossRef全文| Google Scholar