유전학의 경계
소개
프로모터는 게놈의 비 코딩 영역에 속하는 핵심 요소입니다. 그들은 주로 유전자의 활성화 또는 억제를 제어합니다. 이들은 유전자의 전사 시작 부위 (TSS) 근처 및 상류에 위치합니다. 유전자의 프로모터 측면 영역에는 다음을 제공하는 단백질에 대한 인식 부위 역할을하는 많은 중요한 짧은 DNA 요소와 모티프 (5 및 15 염기 길이)가 포함될 수 있습니다. 다운 스트림 유전자의 전사의 적절한 개시 및 조절 (Juven-Gershon et al., 2008). 유전자 전 사체의 개시는 유전자 발현 조절의 가장 기본적인 단계입니다. 프로모터 코어는 TSS를 연결하고 전사를 직접 시작하기에 충분한 DNA 서열의 최소 스트레치입니다. 코어 프로모터의 길이는 일반적으로 60 ~ 120 염기쌍 (bp) 사이입니다.
TATA-box는 전사가 시작되는 다른 분자를 나타내는 프로모터 하위 서열입니다. 반복되는 T와 A 염기쌍 (TATAAA)이 특징이기 때문에 “TATA-box”로 명명되었습니다 (Baker et al., 2003). TATA-box에 대한 대부분의 연구는 인간, 효모, 그러나 Drosophila 게놈은 고세균과 고대 진핵 생물과 같은 다른 종에서도 유사한 요소가 발견되었습니다 (Smale and Kadonaga, 2003). 인간의 경우 유전자의 24 %가 TATA-box를 포함하는 프로모터 영역을 가지고 있습니다 (Yang et al., 2007 진핵 생물에서 TATA-box는 TSS의 상류 ~ 25 bp에 위치합니다 (Xu et al., 2016). 전사 방향을 정의 할 수 있으며 읽을 DNA 가닥을 표시 할 수도 있습니다. TATA-box를 포함한 여러 비 코딩 영역에 결합하고 DNA에서 RNA를 합성하는 RNA 중합 효소라는 효소를 동원합니다.
유전자 전사에서 프로모터의 중요한 역할로 인해 프로모터 부위의 정확한 예측이 가능해집니다. 유전자 발현, 패턴 해석, 구축 및 이해에 필요한 단계 유전자 조절 네트워크의 기능. 돌연변이 분석 (Matsumine et al., 1998) 및 면역 침전 분석 (Kim et al., 2004; Dahl and Collas, 2008)과 같은 프로모터 식별을위한 다양한 생물학적 실험이있었습니다. 그러나 이러한 방법은 비용과 시간이 많이 소요되었습니다. 최근 NGS (Next-generation Sequencing) (Behjati and Tarpey, 2013)의 개발과 함께 다양한 유기체의 더 많은 유전자가 시퀀싱되었으며 그 유전자 요소가 계산적으로 탐구되었습니다 (Zhang et al., 2011). 반면에 NGS 기술의 혁신으로 전체 게놈 시퀀싱 비용이 급격히 감소하여 더 많은 시퀀싱 데이터를 사용할 수 있습니다. 데이터 가용성은 연구자들이 프로모터 예측 작업을위한 계산 모델을 개발하도록 유도합니다. 그러나 여전히 불완전한 작업이며 발기인을 정확하게 예측할 수있는 효율적인 소프트웨어가 없습니다.
발기인 예측자는 활용 된 접근 방식을 기반으로 신호 기반 접근 방식, 콘텐츠 기반 접근 방식의 세 그룹으로 분류 할 수 있습니다. 및 GpG 기반 접근 방식입니다. 신호 기반 예측자는 RNA 중합 효소 결합 부위와 관련된 프로모터 요소에 초점을 맞추고 서열의 비 요소 부분을 무시합니다. 그 결과 예측 정확도가 약하고 만족스럽지 못했습니다. 신호 기반 예측 변수의 예는 다음과 같습니다. PromoterScan (Prestridge, 1995)은 TATA-box의 추출 된 특징과 선형 판별 자와 함께 전사 인자 결합 부위의 가중 매트릭스를 사용하여 프로모터 서열이 아닌 프로모터 서열을 분류합니다. Promoter2.0 (Knudsen, 1999)은 TATA-Box, CAAT-Box 및 GC-Box와 같은 다른 상자에서 기능을 추출하여 분류를 위해 인공 신경망 (ANN)에 전달했습니다. 특징 추출을 위해 Inr (Inritiator Element)와 TATA-Box, 분류를 위해 시간 지연 신경망을 활용 한 NNPP2.1 (Reese, 2001)과 TATA-Box를 활용하고 관련성 벡터 머신을 활용 한 Down and Hubbard (2002) (RVM)을 분류 자로 사용합니다. 콘텐츠 기반 예측자는 시퀀스에서 k- 길이 창을 실행하여 k-mer의 빈도를 계산하는 데 의존했습니다. 그러나 이러한 방법은 시퀀스에서 기본 쌍의 공간 정보를 무시합니다. 콘텐츠 기반 예측 자의 예는 다음과 같습니다. PromFind (Hutchinson, 1996)는 k-mer 빈도를 사용하여 헥사 머 프로모터 예측을 수행합니다. PromoterInspector (Scherf et al., 2000)는 가변 길이 모티프로 정의 된 특정 특징을 스캔하여 중합 효소 II 프로모터의 일반적인 게놈 컨텍스트를 기반으로 프로모터를 포함하는 영역을 식별합니다. MCPromoter1.1 (Ohler et al., 1999)은 프로모터 서열을 예측하기 위해 5 차의 단일 보간 된 Markov 사슬 (IMC)을 사용했습니다.마지막으로, GpG 기반 예측 변수는 GpG 섬의 위치를 프로모터 영역으로 활용했거나 인간 유전자의 첫 번째 엑손 영역은 일반적으로 GpG 섬을 포함합니다 (Ioshikhes and Zhang, 2000; Davuluri et al., 2001; Lander et al., 2001; Ponger와 Mouchiroud, 2002). 그러나 프로모터의 60 %만이 GpG 아일랜드를 포함하므로 이러한 종류의 예측 자의 예측 정확도는 60 %를 초과하지 않았습니다.
최근에는 프로모터 예측에 시퀀스 기반 접근 방식이 사용되었습니다. Yang et al. (2017)은 인핸서-프로모터 상호 작용을 예측하기 위해 가장 관련성이 높은 시퀀스 정보를 캡처하기 위해 다양한 특징 추출 전략을 사용했습니다. Lin et al. (2017)은 원핵 생물에서 sigma70 프로모터의 식별을 위해 “iPro70-PseZNC”라는 서열 기반 예측자를 제안했습니다. 마찬가지로 Bharanikumar et al. (2018)은 서열이 위치 가중치 행렬 (PWM)로 표시되는 동적 다중 회귀 접근법을 기반으로 대장균 프로모터의 강도를 예측하기 위해 PromoterPredict를 제안했습니다. Kanhere와 Bansal (2005)은 프로모터와 비 프로모터 서열을 구별하기 위해 DNA 서열 안정성의 차이를 활용했습니다. Xiao et al. (2018)은 프로모터 서열 식별을위한 iPSW (2L) -PseKNC라는 2 계층 예측 변수를 도입했으며 서열에서 하이브리드 특징을 추출하여 프로모터의 강도를 확인했습니다.
앞서 언급 한 모든 예측 변수에는 도메인이 필요합니다. 기능을 직접 제작하기위한 지식. 반면에 딥 러닝 기반 접근 방식을 사용하면 원시 데이터 (DNA / RNA 시퀀스)를 직접 사용하여보다 효율적인 모델을 구축 할 수 있습니다. 심층 컨볼 루션 신경망은 이미지, 비디오, 오디오 및 음성 처리와 같은 까다로운 작업에서 최첨단 결과를 달성했습니다 (Krizhevsky et al., 2012; LeCun et al., 2015; Schmidhuber, 2015; Szegedy et al. , 2015). 또한 DeepBind (Alipanahi et al., 2015), DeepCpG (Angermueller et al., 2017), 분기점 선택 (Nazari et al., 2018), 대체 접합 부위 예측 (Oubounyt)과 같은 생물학적 문제에 성공적으로 적용되었습니다. et al., 2018), 2 “-O 메틸화 부위 예측 (Tahir et al., 2018), DNA 서열 정량화 (Quang and Xie, 2016), 인간 단백질 subcellular 국소화 (Wei et al., 2018) 등. CNN은 최근 프로모터 인식 작업에서 큰 주목을 받았습니다. 최근 Umarov와 Solovyev (2017)는 짧은 프로모터 서열 식별을위한 CNNprom을 도입했으며,이 CNN 기반 아키텍처는 프로모터 및 비 프로모터 서열 분류에서 높은 결과를 얻었습니다. 그 후이 모델이 개선되었습니다. 저자가 가장 중요한 프로모터 시퀀스 요소를 검사하기 위해 SVM (support vector machine) 분류기를 사용한 Qian et al. (2018)에 의해 다음으로 가장 영향력있는 요소는 압축되지 않은 상태로 유지되고 덜 중요한 요소는 압축됩니다. 이 프로세스는 더 나은 성능을 가져 왔습니다. 최근 Umarov et al.에 의해 긴 프로모터 식별 모델이 제안되었습니다. (2019)에서 저자는 TSS 위치 식별에 초점을 맞췄습니다.
위에 언급 된 모든 작업에서 네거티브 세트는 게놈의 비 프로모터 영역에서 추출되었습니다. 프로모터 염기 서열은 –30 ~ –25bp에 위치한 TATA-box, –110 ~ –80bp에 위치한 GC-Box, –에 위치한 CAAT-Box와 같은 특정 기능적 요소로만 풍부함을 알고 있습니다. 80 ~ -70 bp 등. 이는 시퀀스 구조 측면에서 양성 샘플과 음성 샘플 사이의 큰 차이로 인해 높은 분류 정확도를 가져옵니다. 또한 분류 작업은 쉽게 달성 할 수 없게됩니다. 예를 들어 CNN 모델은 시퀀스 유형을 결정하기 위해 특정 위치에 일부 모티프가 있는지 여부에 의존합니다. 따라서 이러한 모델은 프로모터 모티프가 있지만 프로모터 서열이 아닌 게놈 서열에서 테스트 될 때 정밀도 / 민감도가 매우 낮습니다 (높은 위양성). 프로모터 영역에 속하는 것보다 게놈에 더 많은 TATAAA 모티프가 있다는 것은 잘 알려져 있습니다. 예를 들어, 인간 염색체 1의 DNA 서열, ftp://ftp.ensembl.org/pub/release-57/fasta/homo_sapiens/dna/는 151656 TATAAA 모티프를 포함합니다. 전체 인간 게놈에서 대략적인 최대 유전자 수보다 많습니다. 이 문제를 설명하기 위해 TATA 상자가있는 비 프로모터 시퀀스에서 이러한 모델을 테스트 할 때 대부분의 시퀀스가 잘못 분류된다는 것을 알 수 있습니다. 따라서 강력한 분류기를 생성하기 위해서는 클래스를 구별하기 위해 분류자가 사용할 특성을 결정하므로 네거티브 세트를 신중하게 선택해야합니다. 이 아이디어의 중요성은 (Wei et al., 2014)와 같은 이전 작업에서 입증되었습니다. 이 작업에서 우리는 주로이 문제를 다루고 이러한 모티프에 대한 모델의 종속성을 줄이기 위해 네거티브 클래스에 포지티브 클래스 기능 모티프의 일부를 통합하는 접근 방식을 제안합니다.LSTM 모델과 결합 된 CNN을 사용하여 인간 및 마우스 TATA 및 비 TATA 진핵 프로모터의 서열 특성을 분석하고 비 프로모터 서열과 짧은 프로모터 서열을 정확하게 구별 할 수있는 계산 모델을 구축합니다.
재료 및 방법
2.1. 데이터 세트
제안 된 프로모터 예측자를 학습하고 테스트하는 데 사용되는 데이터 세트는 사람과 마우스에서 수집됩니다. 이들은 프로모터의 두 가지 다른 부류, 즉 TATA 프로모터 (즉, TATA- 박스를 포함하는 서열) 및 비 -TATA 프로모터를 포함합니다. 이러한 데이터 세트는 Eukaryotic Promoter Database (EPDnew)에서 구축되었습니다 (Dreos et al., 2012). EPDnew는 잘 알려진 EPD 데이터 세트 (Périer et al., 2000)의 새로운 섹션으로, 전사 시작 부위가 실험적으로 결정된 진핵 POL II 프로모터의 중복되지 않은 컬렉션에 주석이 추가되었습니다. ENSEMBL 프로모터 컬렉션 (Dreos et al., 2012)과 비교하여 고품질 프로모터를 제공하며 https://epd.epfl.ch//index.php에서 공개적으로 액세스 할 수 있습니다. 우리는 EPDnew에서 각 유기체에 대한 TATA 및 비 -TATA 프로모터 게놈 서열을 다운로드했습니다. 이 작업으로 Human-TATA, Human-non-TATA, Mouse-TATA 및 Mouse-non-TATA의 네 가지 프로모터 데이터 세트를 얻었습니다. 이러한 각 데이터 세트에 대해 다음 섹션에서 설명하는 제안 된 접근 방식을 기반으로 동일한 크기의 포지티브 세트 (프로모터가 아닌 시퀀스)가 구성됩니다. 각 유기체의 프로모터 염기 서열 수에 대한 자세한 내용은 표 1에 나와 있습니다. 모든 염기 서열은 길이가 300bp이며 -249 ~ + 50bp (+1은 TSS 위치를 나타냄)에서 추출되었습니다. 품질 관리로서 제안 된 모델을 평가하기 위해 5 겹 교차 검증을 사용했습니다. 이 경우 3 겹은 훈련에 사용되고 1 겹은 검증에 사용되고 나머지 접기는 테스트에 사용됩니다. 따라서 제안 된 모델은 5 번 학습되고 5 배의 전체 성능이 계산됩니다.

표 1.이 연구에 사용 된 네 가지 데이터 세트의 통계.
2.2. Negative Dataset Construction
프로모터 및 비 프로모터 시퀀스 분류를 정확하게 수행 할 수있는 모델을 훈련하려면 네거티브 세트 (비 프로모터 시퀀스)를 신중하게 선택해야합니다. 이 점은 모델을 잘 일반화 할 수 있도록 만드는 데 중요하므로 더 까다로운 데이터 세트에서 평가할 때 정밀도를 유지할 수 있습니다. (Qian et al., 2018)과 같은 이전 작품은 게놈 비 프로모터 영역에서 무작위로 조각을 선택하여 네거티브 세트를 구성했습니다. 분명히이 접근법은 양수 세트와 음수 세트 사이에 교차점이 없기 때문에 완전히 합리적이지 않습니다. 따라서 모델은 두 클래스를 분리하는 기본 기능을 쉽게 찾을 수 있습니다. 예를 들어, TATA 모티프는 특정 위치의 모든 양성 서열에서 찾을 수 있습니다 (일반적으로 TSS의 업스트림 28bp, 데이터 세트에서 –30에서 –25 pb 사이). 따라서이 모티프를 포함하지 않는 네거티브 세트를 무작위로 생성하면이 데이터 세트에서 높은 성능을 얻을 수 있습니다. 그러나이 모델은 TATA 모티프를 프로모터로 갖는 음성 서열을 분류하는 데 실패합니다. 간단히 말해서,이 접근 방식의 주요 결함은 딥 러닝 모델을 훈련 할 때 특정 위치에 몇 가지 간단한 기능이 있는지 여부에 따라 긍정 및 부정 클래스를 구별하는 방법 만 배우므로 이러한 모델을 실행 불가능하게 만든다는 것입니다. 이 작업에서 우리는 긍정 세트에서 부정 세트를 도출하는 대체 방법을 설정하여이 문제를 해결하는 것을 목표로합니다.
우리의 방법은 특성이 부정과 특성 사이에 공통적이라는 사실에 기반을두고 있습니다. 포지티브 클래스 모델은 결정을 내릴 때 이러한 기능에 대한 종속성을 무시하거나 줄이는 경향이 있습니다 (즉, 이러한 기능에 낮은 가중치를 할당). 대신 모델은 더 깊고 덜 분명한 특징을 검색해야합니다. 딥 러닝 모델은 일반적으로 이러한 유형의 데이터를 학습하는 동안 느린 수렴으로 어려움을 겪습니다. 그러나이 방법은 모델의 견고성을 향상시키고 일반화를 보장합니다. 다음과 같이 네거티브 세트를 재구성합니다. 각 양수 시퀀스는 하나의 음수 시퀀스를 생성합니다. 양성 시퀀스는 20 개의 하위 시퀀스로 나뉩니다. 그런 다음 12 개의 하위 시퀀스가 무작위로 선택되고 무작위로 대체됩니다. 나머지 8 개의 하위 시퀀스는 보존됩니다. 이 프로세스는 그림 1에 설명되어 있습니다.이 프로세스를 포지티브 세트에 적용하면 프로모터 시퀀스 (변경되지 않은 하위 시퀀스, 20 개 중 8 개 하위 시퀀스)에서 보존 된 부분이있는 새로운 비 프로모터 시퀀스가 생성됩니다. 이들 매개 변수는 프로모터 서열의 보존 된 부분을 포함하는 서열의 32 및 40 %를 갖는 네거티브 세트를 생성 할 수있게한다. 이 비율은 섹션 3.2에 설명 된대로 강력한 프로모터 예측자를 갖는 데 최적 인 것으로 밝혀졌습니다.보존 된 부분이 네거티브 시퀀스에서 동일한 위치를 차지하기 때문에 TATA-box 및 TSS와 같은 명백한 모티프는 이제 32 ~ 40 %의 비율로 두 세트간에 공통적입니다. 인간 및 마우스 TATA 프로모터 데이터에 대한 양성 및 음성 세트의 서열 로고는 각각 그림 2, 3에 나와 있습니다. -30 및 -25 bp 위치의 TATA 모티프와 +1 bp 위치의 TSS와 같이 동일한 위치에서 포지티브 및 네거티브 세트가 동일한 기본 모티프를 공유 함을 알 수 있습니다. 따라서 학습이 더 어렵지만 결과 모델은 잘 일반화됩니다.
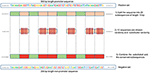
그림 1. 네거티브 세트 구성 방법의 예시. 녹색은 임의로 보존 된 하위 시퀀스를 나타내고 빨간색은 임의로 선택 및 대체 된 시퀀스를 나타냅니다.

그림 2. 양성 집합 (A) 및 음성 집합 (B) 모두에 대한 인간 TATA 프로모터의 시퀀스 로고. 플롯은 두 세트 사이의 기능적 모티프의 보존을 보여줍니다.

그림 3. 양성 집합 (A) 및 음성 집합 (B) 모두에 대한 마우스 TATA 프로모터의 시퀀스 로고. 플롯은 두 세트 사이의 기능적 모티프의 보존을 보여줍니다.
2.3. 제안 된 모델
그림 4와 같이 컨볼 루션 레이어와 반복 레이어를 결합하는 딥 러닝 모델을 제안합니다. 단일 원시 게놈 시퀀스 S = {N1, N2,…, Nl}을 허용합니다. 여기서 N ∈ {A, C, G, T} 및 l은 입력 시퀀스의 길이이며 실수 값 점수를 입력하고 출력합니다. 입력은 원-핫 인코딩되고 4 개의 채널이있는 1 차원 벡터로 표시됩니다. 벡터 l = 300의 길이와 4 개 채널은 A, C, G, T이며 (1 0 0 0), (0 1 0 0), (000 1 0), (000 1 )입니다. 가장 성능이 좋은 모델을 선택하기 위해 그리드 검색 방법을 사용하여 최상의 하이퍼 매개 변수를 선택했습니다. 우리는 CNN 단독, LSTM 단독, BiLSTM 단독, LSTM과 결합 된 CNN과 같은 다른 아키텍처를 시도했습니다. 조정 된 하이퍼 파라미터는 컨볼 루션 계층의 수, 커널 크기, 각 계층의 필터 수, 최대 풀링 계층의 크기, 드롭 아웃 확률 및 Bi-LSTM 계층의 단위입니다.
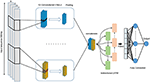
그림 4. 제안 된 DeePromoter 모델의 아키텍처
제안 된 모델은 병렬로 정렬 된 다중 컨볼 루션 레이어로 시작하여 다른 창 크기를 가진 입력 시퀀스의 중요한 모티프를 학습하는 데 도움이됩니다. 창 크기가 27, 14, 7 인 비 TATA 프로모터에 대해 3 개의 컨볼 루션 레이어를 사용하고, 창 크기가 27, 14 인 TATA 프로모터에 대해 2 개의 컨볼 루션 레이어를 사용합니다. 모든 컨볼 루션 레이어는 ReLU 활성화 함수 다음에 따릅니다 (Glorot et al. , 2011), 창 크기가 6 인 최대 풀링 레이어 및 확률 0.5의 드롭 아웃 레이어. 그런 다음, 이러한 계층의 출력은 함께 연결되어 회선 계층에서 학습 된 모티프 간의 종속성을 캡처하기 위해 32 개의 노드가있는 양방향 장단기 기억 (BiLSTM) (Schuster and Paliwal, 1997) 계층에 공급됩니다. BiLSTM 이후 학습 된 기능은 평평 해지고 0.5 확률로 드롭 아웃이 이어집니다. 그런 다음 분류를 위해 두 개의 완전 연결 계층을 추가합니다. 첫 번째 계층에는 128 개의 노드가 있고 그 다음에는 0.5의 확률로 ReLU 및 드롭 아웃이 뒤 따르고 두 번째 계층은 하나의 노드와 시그 모이 드 활성화 함수가있는 예측에 사용됩니다. BiLSTM를 사용하면 정보가 DNA 및 RNA와 같은 순차적 샘플의 장기적인 종속성을 유지하고 학습 할 수 있습니다. 이는 메모리 셀과 입력, 출력, 잊어 버린 게이트라고하는 3 개의 게이트로 구성된 LSTM 구조를 통해 이루어집니다. 이 게이트는 메모리 셀의 정보를 조절하는 역할을합니다. 또한 LSTM 모듈을 사용하면 네트워크 깊이가 증가하고 필요한 매개 변수 수가 적습니다. 네트워크가 더 깊어지면 더 복잡한 특징을 추출 할 수 있으며, 네거티브 세트에 하드 샘플이 포함되어 있으므로 이것이 모델의 주요 목표입니다.
Kera 프레임 워크는 제안 된 모델을 구성하고 훈련하는 데 사용됩니다 (Chollet F. et al., 2015). Adam Optimizer (Kingma and Ba, 2014)는 학습률이 0.001 인 매개 변수를 업데이트하는 데 사용됩니다. 배치 크기는 32로 설정되고 Epoch 수는 50으로 설정됩니다. 조기 중지는 유효성 검사 손실을 기반으로 적용됩니다.
결과 및 논의
3.1. 성능 측정
이 작업에서는 제안 된 모델의 성능을 평가하기 위해 널리 채택 된 평가 측정 항목을 사용합니다.이러한 측정 항목은 정밀도, 재현율 및 매튜 상관 계수 (MCC)이며 다음과 같이 정의됩니다.
TP가 참 양성이고 올바르게 식별 된 프로모터 서열을 나타내는 경우, TN은 참 음성이고 올바르게 거부 된 프로모터 서열을 나타내고, FP는 거짓 양성이며 잘못 식별됨을 나타냅니다. 프로모터 서열 및 FN은 위음성이며 잘못 거부 된 프로모터 서열을 나타냅니다.
3.2. 네거티브 세트의 효과
프로모터 서열 식별을 위해 이전에 출판 된 작업을 분석 할 때 우리는 이러한 작업의 성능이 네거티브 데이터 세트를 준비하는 방법에 크게 좌우된다는 것을 발견했습니다. 그들은 준비한 데이터 세트에서 매우 잘 수행했지만, 프로모터 시퀀스가있는 공통 모티프를 갖는 비 프롬프터 시퀀스를 포함하는 더 까다로운 데이터 세트에서 평가할 때 높은 위양성 비율을 나타냅니다. 예를 들어, TATA 프로모터 데이터 세트의 경우 무작위로 생성 된 시퀀스는 -30 및 -25 bp 위치에 TATA 모티프가 없으므로 분류 작업이 더 쉬워집니다. 즉, 분류자는 프로모터 서열을 식별하기 위해 TATA 모티프의 존재에 의존했으며 결과적으로 그들이 준비한 데이터 세트에서 높은 성능을 쉽게 달성 할 수있었습니다. 그러나 그들의 모델은 TATA 모티프를 포함하는 네거티브 시퀀스를 다룰 때 극적으로 실패했습니다 (하드 예제). 오 탐률이 증가함에 따라 정밀도가 떨어졌습니다. 간단히 말해서, 그들은 이러한 서열을 양성 프로모터 서열로 분류했습니다. 다른 프로모터 모티프에 대해서도 유사한 분석이 유효합니다. 따라서 우리 작업의 주요 목적은 특정 데이터 세트에서 고성능을 달성 할뿐만 아니라 까다로운 데이터 세트에 대한 학습을 통해 잘 일반화하는 모델 능력을 향상시키는 것입니다.
이 점을 더 설명하기 위해 우리는 네거티브 세트 준비의 다른 방법으로 인간 및 마우스 TATA 프로모터 데이터 세트에서 모델을 테스트하십시오. 첫 번째 실험은 게놈의 비 코딩 영역에서 무작위로 샘플링 된 음성 시퀀스를 사용하여 수행됩니다 (즉, 이전 작업에서 사용 된 접근 방식과 유사 함). 놀랍게도, 우리가 제안한 모델은 인간과 마우스 모두에 대해 각각 거의 완벽한 예측 정확도 (정밀도 = 99 %, 재현율 = 99 %, Mcc = 98 %) 및 (정밀도 = 99 %, 재현율 = 98 %, Mcc = 97 %)를 달성했습니다. . 이러한 높은 결과가 예상되지만 문제는이 모델이 어려운 예제가있는 데이터 세트에서 평가할 때 동일한 성능을 유지할 수 있는지 여부입니다. 이전 모델을 분석 한 결과 대답은 ‘아니요’입니다. 두 번째 실험은 2.2 절에서 설명한대로 데이터 세트를 준비하기 위해 제안 된 방법을 사용하여 수행됩니다. 12, 20, 32 및 40 %와 같은 다른 비율로 보존 된 TATA 상자를 포함하는 네거티브 세트를 준비하고 목표는 정밀도와 리콜 사이의 간격을 줄이는 것입니다. 이를 통해 우리 모델은 TATA-box의 유무 만 배우는 것이 아니라 더 복잡한 기능을 학습 할 수 있습니다. 그림 5A, B에서 볼 수 있듯이 모델은 인간 및 마우스 TATA 프로모터 데이터 세트 모두에 대해 32 ~ 40 %의 비율로 안정화됩니다.
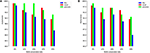
그림 5. 인간 (A) 및 마우스 (B) 모두에 대한 TATA 프로모터 데이터 세트의 경우 음수 세트에서 TATA 모티프의 다양한 보존 비율이 성능에 미치는 영향 .
3.3. 결과 및 비교
지난 몇 년 동안 많은 프로모터 지역 예측 도구가 제안되었습니다 (Hutchinson, 1996; Scherf et al., 2000; Reese, 2001; Umarov 및 Solovyev, 2017). 그러나 이러한 도구 중 일부는 테스트에 공개적으로 사용할 수 없으며 일부 도구는 원시 게놈 서열 외에 더 많은 정보가 필요합니다. 이 연구에서는 제안 된 모델의 성능을 표 2와 같이 Umarov와 Solovyev (2017)가 제안한 최신 최신 작업 인 CNNProm과 비교합니다. 일반적으로 제안 된 모델 인 DeePromoter, 모든 평가 지표를 사용하여 모든 데이터 세트에서 CNNProm을 분명히 능가합니다. 보다 구체적으로 DeePromoter는 인간 TATA 데이터 세트의 경우 정밀도, 재현율 및 MCC를 각각 0.18, 0.04 및 0.26 향상시킵니다. 인간이 아닌 TATA 데이터 세트의 경우 DeePromoter는 정밀도를 0.39, 재현율을 0.12, MCC를 0.66 개선합니다. 마찬가지로 DeePromoter는 마우스 TATA 데이터 세트의 경우 정밀도를 각각 0.24 및 0.31 씩 향상시킵니다. 마우스가 TATA가 아닌 데이터 세트의 경우 DeePromoter는 정밀도를 0.37, 재현율을 0.04, MCC를 0.65 향상시킵니다. 이러한 결과는 CNNProm이 TATA 프로모터로 음성 서열을 거부하지 못하여 높은 위양성을 가지고 있음을 확인합니다. 반면에 우리 모델은 이러한 사례를 더 성공적으로 처리 할 수 있으며 CNNProm에 비해 오 탐률이 낮습니다.

표 2. DeePromoter와 상태 비교 -the-art method.
추가 분석을 위해 출력 점수의 각 위치에서 뉴클레오티드가 번갈아 나타나는 효과를 연구합니다. 우리는 프로모터 서열의 가장 중요한 부분을 호스팅하는 영역 –40 및 10bp에 중점을 둡니다. 테스트 세트의 각 프로모터 시퀀스에 대해 컴퓨터 돌연변이 스캐닝을 수행하여 입력 하위 시퀀스의 모든 염기 돌연변이 효과를 평가합니다 (-40 ~ 10 bp 하위 시퀀스 간격에 150 개의 치환). 이것은 인간 및 마우스 TATA 데이터 세트에 대해 각각 그림 6, 7에 설명되어 있습니다. 파란색은 변이로 인한 출력 점수의 하락을 나타내고 빨간색은 변이로 인한 점수의 증가를 나타냅니다. -30 및 -25 bp 영역에서 뉴클레오티드를 C 또는 G로 변경하면 출력 점수가 크게 감소합니다. 이 영역은 프로모터 서열에서 매우 중요한 기능적 모티프 인 TATA-box입니다. 따라서 우리 모델은이 지역의 중요성을 성공적으로 찾을 수 있습니다. 나머지 위치에서, 특히 마우스의 경우 C 및 G 뉴클레오티드가 A 및 T보다 더 바람직합니다. 이것은 프로모터 영역이 A와 T보다 더 많은 C와 G 뉴클레오티드를 가지고 있다는 사실로 설명 할 수 있습니다 (Shi and Zhou, 2006).

그림 6. 인간 TATA 프로모터 서열의 경우 TATA 상자를 포함하는 –40bp ~ 10bp 영역의 돌출 맵

그림 7. 마우스 TATA 프로모터 시퀀스의 경우 TATA 상자를 포함하는 –40bp ~ 10bp 영역의 돌출 맵
결론
유전자 조절 과정의 기본 메커니즘을 이해하려면 프로모터 서열의 정확한 예측이 필수적입니다. 이 연구에서 우리는 TATA 및 비 TATA 프로모터 모두에 대해 인간과 마우스의 경우 짧은 진핵 프로모터 서열을 예측하기 위해 컨볼 루션 신경망과 양방향 LSTM의 조합을 기반으로하는 DeePromoter를 개발했습니다. 이 작업의 필수 구성 요소는 프로모터 및 비 프로모터 시퀀스를 분류 할 때 시퀀스의 몇 가지 명백한 특징 / 모티프에 대한 의존으로 인해 이전에 개발 된 도구에서 발견 된 낮은 정밀도 (높은 위양성 비율) 문제를 극복하는 것이 었습니다. 이 작업에서 우리는 일부 기능적 모티프의 존재를 기반으로 프로모터와 비 프로모터 시퀀스를 구별하는 대신 모델이 깊고 관련성있는 시퀀스를 탐색하도록 유도하는 하드 네거티브 세트를 구성하는 데 특히 관심이있었습니다. DeePromoter 사용의 주요 이점은 까다로운 데이터 세트에서 높은 정확도를 달성하면서 오 탐지 예측 수를 크게 줄인다는 것입니다. DeePromoter는 성능뿐만 아니라 높은 오 탐지 예측 문제를 극복하는 데있어 이전 방법보다 성능이 뛰어났습니다. 이 프레임 워크는 약물 관련 응용 프로그램 및 학계에 도움이 될 것으로 예상됩니다.
저자 기고
MO와 ZL은 데이터 세트를 준비하고 알고리즘을 구상하고 실험을 수행하고 분석. MO와 HT는 웹 서버를 준비하고 ZL과 KC의 지원을 받아 원고를 작성했습니다. 모든 저자가 결과를 논의하고 최종 원고에 기여했습니다.
기금
이 연구는 한국 정부 (National Research Foundation)의 뇌 연구 프로그램 (NRF)의 지원을 받았습니다. MSIT) (No. NRF-2017M3C7A1044815).
이해 상충 성명
저자는 연구가 다음과 같이 해석 될 수있는 상업적 또는 재정적 관계가없는 상태에서 수행되었다고 선언합니다. 잠재적 이해 상충.
Bharanikumar, R., Premkumar, KAR 및 Palaniappan, A. (2018). 프로모터 예측 : 대장균 σ70 프로모터 강도의 서열 기반 모델링은 프로모터 강도와 서열 사이의 대수 의존성을 산출합니다. PeerJ 6 : e5862. doi : 10.7717 / peerj.5862
PubMed 요약 | CrossRef 전체 텍스트 | Google 학술 검색
Glorot, X., Bordes, A. 및 Bengio, Y. (2011). “Deep sparse rectifier neural networks”, Proceedings of the Fourteenth International Conference on Artificial Intelligence and Statistics, (Fort Lauderdale, FL 🙂 315–323.
Google Scholar
Hutchinson, G. (1996). 차동 6 량체 주파수 분석을 사용한 척추 동물 프로모터 영역 예측. Bioinformatics 12, 391–398.
PubMed Abstract | Google Scholar
Kingma, DP 및 Ba, J. (2014). Adam : 확률 적 최적화 방법. arXiv preprint arXiv : 1412.6980.
Google Scholar
Knudsen, S. (1999). Promoter2. 0 : polii 프로모터 서열 인식 용. Bioinformatics 15, 356–361.
PubMed 요약 | Google 학술 검색
Ponger, L. 및 Mouchiroud, D. (2002). Cpgprod : 큰 게놈 포유류 서열에서 전사 시작 부위와 관련된 cpg 섬 식별. 생물 정보학 18, 631–633. doi : 10.1093 / bioinformatics / 18.4.631
PubMed 요약 | CrossRef 전체 텍스트 | Google 학술 검색
Quang, D. 및 Xie, X. (2016). Danq : DNA 시퀀스의 기능을 정량화하기위한 하이브리드 컨볼 루션 및 반복 심층 신경망. Nucleic Acids Res. 44, e107–e107. doi : 10.1093 / nar / gkw226
PubMed 요약 | CrossRef 전체 텍스트 | Google 학술 검색
Umarov, R.K. 및 Solovyev, V. V. (2017). 컨볼 루션 딥 러닝 신경망을 사용하여 원핵 및 진핵 프로모터의 인식. PLoS ONE 12 : e0171410. doi : 10.1371 / journal.pone.0171410
PubMed 요약 | CrossRef 전체 텍스트 | Google 학술 검색