Grenser i genetikk
Innledning
Arrangører er nøkkelelementene som tilhører ikke-kodende regioner i genomet. De styrer i stor grad aktivering eller undertrykkelse av genene. De er lokalisert nær og oppstrøms genets transkripsjonsstartsted (TSS). Et gen som promoterflankerende region kan inneholde mange viktige korte DNA-elementer og motiver (5 og 15 baser lange) som fungerer som gjenkjenningssteder for proteiner som gir riktig initiering og regulering av transkripsjon av nedstrømsgenet (Juven-Gershon et al., 2008). Initiering av gentranskripsjon er det mest grunnleggende trinnet i reguleringen av genuttrykk. Promotorkjernen er en minimal strekning av DNA-sekvens som kiler TSS og er tilstrekkelig til å direkte starte transkripsjonen. Lengden på kjernepromotoren varierer vanligvis mellom 60 og 120 basepar (bp).
TATA-boksen er en promotorsekvens som indikerer for andre molekyler hvor transkripsjon begynner. Den ble kalt «TATA-box» da dens sekvens er preget av å gjenta T- og A-basepar (TATAAA) (Baker et al., 2003). De aller fleste studier på TATA-boksen har blitt utført på mennesker, gjær, og Drosophila genomer, men lignende elementer har blitt funnet i andre arter som archaea og eldgamle eukaryoter (Smale og Kadonaga, 2003). I menneskelige tilfeller har 24% av gener promoterregioner som inneholder TATA-box (Yang et al., 2007 I eukaryoter er TATA-box lokalisert til ~ 25 bp oppstrøms for TSS (Xu et al., 2016). Den er i stand til å definere transkripsjonsretningen og indikerer også DNA-strengen som skal leses. Proteiner kalt transkripsjonsfaktorer binde seg til flere ikke-kodende regioner inkludert TATA-box og rekruttere et enzym kalt RNA-polymerase, som syntetiserer RNA fra DNA.
På grunn av den viktige rollen promotorene har i gentranskripsjon, blir nøyaktig prediksjon av promotersteder et nødvendig trinn i genuttrykk, fortolkning av mønstre, og å bygge og forstå funksjonaliteten til genetiske reguleringsnettverk. Det var forskjellige biologiske eksperimenter for identifikasjon av promotorer som mutasjonsanalyse (Matsumine et al., 1998) og immunutfellingsanalyser (Kim et al., 2004; Dahl og Collas, 2008). Imidlertid var disse metodene både dyre og tidkrevende. Nylig, med utviklingen av neste generasjons sekvensering (NGS) (Behjati og Tarpey, 2013), har flere gener fra forskjellige organismer blitt sekvensert og genelementene deres blitt utforsket (Zhang et al., 2011). På den annen side har innovasjonen av NGS-teknologi resultert i et dramatisk fall i kostnadene for hele genom-sekvenseringen, og dermed er flere sekvenseringsdata tilgjengelig. Datatilgjengeligheten tiltrekker forskere til å utvikle beregningsmodeller for promotor prediksjon oppgave. Imidlertid er det fortsatt en ufullstendig oppgave, og det er ingen effektiv programvare som nøyaktig kan forutsi promotorer.
Promotorprediktorer kan kategoriseres basert på den benyttede tilnærmingen i tre grupper, nemlig signalbasert tilnærming, innholdsbasert tilnærming og den GpG-baserte tilnærmingen. Signalbaserte prediktorer fokuserer på promoterelementer relatert til RNA-polymerase-bindingssted og ignorerer ikke-elementdelene av sekvensen. Som et resultat var spådomsnøyaktigheten svak og ikke tilfredsstillende. Eksempler på signalbaserte prediktorer inkluderer: PromoterScan (Prestridge, 1995) som brukte de ekstraherte egenskapene til TATA-boksen og en vektet matrise av transkripsjonsfaktorbindingsseter med en lineær diskriminator for å klassifisere promotorsekvenser fra ikke-promoter; Promoter2.0 (Knudsen, 1999) som hentet ut funksjonene fra forskjellige bokser som TATA-Box, CAAT-Box og GC-Box og sendte dem til kunstige nevrale nettverk (ANN) for klassifisering; NNPP2.1 (Reese, 2001) som benyttet initiatorelement (Inr) og TATA-Box for ekstraksjon av funksjoner og et tidsforsinket nevralt nettverk for klassifisering, og Down and Hubbard (2002) som brukte TATA-Box og brukte en relevansvektormaskiner (RVM) som klassifikator. Innholdsbaserte prediktorer stolte på å telle frekvensen av k-mer ved å kjøre et vindu med k-lengde over sekvensen. Imidlertid ignorerer disse metodene den romlige informasjonen til baseparene i sekvensene. Eksempler på innholdsbaserte prediktorer inkluderer: PromFind (Hutchinson, 1996) som brukte k-mer-frekvensen til å utføre heksamer-promoter-prediksjon; PromoterInspector (Scherf et al., 2000) som identifiserte regionene som inneholder promotere basert på en felles genomisk kontekst av polymerase II-promotorer ved å skanne etter spesifikke egenskaper definert som motiv med variabel lengde; MCPromoter1.1 (Ohler et al., 1999) som brukte en enkelt interpolert Markov-kjede (IMC) av 5. orden for å forutsi promotorsekvenser.Til slutt brukte GpG-baserte prediktorer lokaliseringen av GpG-øyene som promoterregion eller den første eksonregionen i de menneskelige gener inneholder vanligvis GpG-øyer (Ioshikhes og Zhang, 2000; Davuluri et al., 2001; Lander et al., 2001; Ponger og Mouchiroud, 2002). Imidlertid inneholder bare 60% av promotorene GpG-øyer, og derfor har prediksjonsnøyaktigheten til denne typen prediktorer aldri overskredet 60%.
Nylig har sekvensbaserte tilnærminger blitt brukt for promotor prediksjon. Yang et al. (2017) benyttet forskjellige funksjonsekstraksjonsstrategier for å fange den mest relevante sekvensinformasjonen for å forutsi interaksjoner mellom forsterkere og promotorer. Lin et al. (2017) foreslo en sekvensbasert prediktor, kalt «iPro70-PseZNC», for sigma70-promoterens identifikasjon i prokaryoten. Likeledes Bharanikumar et al. (2018) foreslo PromoterPredict for å forutsi styrken til Escherichia coli-promotorer basert på en dynamisk multippel regresjonsmetode der sekvensene ble representert som posisjonsvektmatriser (PWM). Kanhere og Bansal (2005) benyttet forskjellene i DNA-sekvensstabilitet mellom promoter- og ikke-promotorsekvenser for å skille dem. Xiao et al. (2018) introduserte en to-lags prediktor kalt iPSW (2L) -PseKNC for identifisering av promotorsekvenser, samt styrken til promotorene ved å trekke ut hybridfunksjoner fra sekvensene.
Alle de ovennevnte prediktorene krever domene- kunnskap for å håndlaget funksjonene. På den annen side gjør dyp læringsbaserte tilnærminger det mulig å bygge mer effektive modeller ved hjelp av rådata (DNA / RNA-sekvenser) direkte. Dypt konvolusjonalt nevrale nettverk oppnådde topp moderne resultater i utfordrende oppgaver som å behandle bilde, video, lyd og tale (Krizhevsky et al., 2012; LeCun et al., 2015; Schmidhuber, 2015; Szegedy et al. , 2015). I tillegg ble den med suksess brukt i biologiske problemer som DeepBind (Alipanahi et al., 2015), DeepCpG (Angermueller et al., 2017), valg av forgreningspunkt (Nazari et al., 2018), alternativ spådomsspådommer (Oubounyt et al., 2018), 2 «-Omethylation sites prediction (Tahir et al., 2018), DNA-sekvenskvantifisering (Quang og Xie, 2016), humant protein subcellulær lokalisering (Wei et al., 2018), etc. Videre, CNN fikk nylig betydelig oppmerksomhet i oppgavens anerkjennelsesoppgave. Umarov og Solovyev (2017) introduserte nylig CNNprom for diskriminering av korte promotorsekvenser, denne CNN-baserte arkitekturen oppnådde høye resultater i å klassifisere promotorsekvenser og ikke-promotersekvenser. av Qian et al. (2018) hvor forfatterne brukte støttevektormaskin (SVM) klassifikator for å inspisere de viktigste promotorsekvenselementene. Deretter ble de mest innflytelsesrike elementene holdt ukomprimert mens de komprimerte de mindre viktige elementene. Denne prosessen resulterte i bedre ytelse. Nylig ble lang promoteridentifikasjonsmodell foreslått av Umarov et al. (2019) der forfatterne fokuserte på identifiseringen av TSS-posisjon.
I alle de ovennevnte verkene ble det negative settet hentet fra ikke-promoterregioner i genomet. Å vite at promotorsekvensene er rike utelukkende av spesifikke funksjonelle elementer som TATA-box som er lokalisert ved –30 ~ –25 bp, GC-Box som ligger ved –110 ~ –80 bp, CAAT-Box som ligger ved – 80 ~ –70 bp, etc. Dette resulterer i høy klassifiseringsnøyaktighet på grunn av stor forskjell mellom de positive og negative prøvene når det gjelder sekvensstruktur. I tillegg blir klassifiseringsoppgaven uanstrengt å oppnå, for eksempel vil CNN-modellene bare stole på tilstedeværelsen eller fraværet av noen motiver i deres spesifikke posisjoner for å ta avgjørelsen om sekvenstypen. Dermed har disse modellene veldig lav presisjon / sensitivitet (høy falsk positiv) når de testes på genomiske sekvenser som har promotormotiver, men de er ikke promotorsekvenser. Det er velkjent at det er flere TATAAA-motiver i genomet enn de som tilhører promoterregionene. For eksempel inneholder DNA-sekvensen til det humane kromosomet 1, ftp://ftp.ensembl.org/pub/release-57/fasta/homo_sapiens/dna/, for eksempel 151 656 TATAAA-motiver. Det er mer enn det omtrentlige maksimale antall gener i det totale humane genomet. Som en illustrasjon på dette problemet merker vi at når vi tester disse modellene på ikke-promotorsekvenser som har TATA-boks, klassifiserer de de fleste av disse sekvensene feil. Derfor, for å generere en robust klassifisering, bør det negative settet velges nøye, da det bestemmer funksjonene som skal brukes av klassifisereren for å diskriminere klassene. Viktigheten av denne ideen er demonstrert i tidligere arbeider som (Wei et al., 2014). I dette arbeidet tar vi hovedsakelig opp dette problemet og foreslår en tilnærming som integrerer noen av de positive klassefunksjonelle motivene i den negative klassen for å redusere modellens avhengighet av disse motivene.Vi bruker en CNN kombinert med LSTM-modell for å analysere sekvensegenskapene til TATA fra mennesker og mus og ikke-TATA eukaryote promotere og bygge beregningsmodeller som nøyaktig kan diskriminere korte promotorsekvenser fra ikke-promoter.
Materialer og metoder
2.1. Datasett
Datasettene, som brukes til å trene og teste den foreslåtte promotoren, er hentet fra mennesker og mus. De inneholder to karakteristiske klasser av promotorene, nemlig TATA-promotorer (dvs. sekvensene som inneholder TATA-box) og ikke-TATA-promotorer. Disse datasettene ble bygget fra Eukaryotic Promoter Database (EPDnew) (Dreos et al., 2012). EPDnew er en ny seksjon under det velkjente EPD-datasettet (Périer et al., 2000) som er kommentert en ikke-redundant samling av eukaryote POL II-promotorer der transkripsjonsstartsted er bestemt eksperimentelt. Det gir promotorer av høy kvalitet sammenlignet med ENSEMBL-promotorsamling (Dreos et al., 2012), og det er offentlig tilgjengelig på https://epd.epfl.ch//index.php. Vi lastet ned TATA og ikke-TATA promoter genomiske sekvenser for hver organisme fra EPDnew. Denne operasjonen resulterte i å oppnå fire promoterdatasett, nemlig: Human-TATA, Human-non-TATA, Mouse-TATA og Mouse-non-TATA. For hvert av disse datasettene konstrueres et negativt sett (ikke-promotorsekvenser) med samme størrelse som det positive basert på den foreslåtte tilnærmingen som beskrevet i neste avsnitt. Detaljene om antall promotorsekvenser for hver organisme er gitt i tabell 1. Alle sekvenser har en lengde på 300 bp og ble ekstrahert fra -249 ~ + 50 bp (+1 refererer til TSS-posisjon). Som kvalitetskontroll brukte vi fem ganger kryssvalidering for å vurdere den foreslåtte modellen. I dette tilfellet brukes 3-fold for trening, 1-fold brukes til validering, og den gjenværende folden brukes til testing. Dermed blir den foreslåtte modellen trent 5 ganger, og den totale ytelsen til 5 ganger beregnes.

Tabell 1. Statistikk over de fire datasettene som er brukt i denne studien.
2.2. Negativ datasettkonstruksjon
For å trene en modell som nøyaktig kan utføre promoter- og ikke-promotersekvensklassifisering, må vi velge negativt sett (ikke-promotersekvenser) nøye. Dette punktet er avgjørende for å lage en modell som er i stand til å generalisere godt, og derfor kan opprettholde sin presisjon når den blir evaluert på mer utfordrende datasett. Tidligere arbeider, som (Qian et al., 2018), konstruerte negative sett ved å tilfeldig velge fragmenter fra ikke-promotorregioner. Åpenbart er denne tilnærmingen ikke helt rimelig, for hvis det ikke er noe skjæringspunkt mellom positive og negative sett. Dermed vil modellen lett finne grunnleggende funksjoner for å skille de to klassene. For eksempel kan TATA-motiv finnes i alle positive sekvenser på en bestemt posisjon (normalt 28 bp oppstrøms for TSS, mellom –30 og –25 pb i vårt datasett). Derfor vil det å skape negativt sett tilfeldig som ikke inneholder dette motivet gi høy ytelse i dette datasettet. Imidlertid klarer ikke modellen å klassifisere negative sekvenser som har TATA-motiv som promotorer. Kort fortalt er den største feilen i denne tilnærmingen at når man trener en dyp læringsmodell, lærer den bare å diskriminere de positive og negative klassene basert på tilstedeværelse eller fravær av noen enkle funksjoner på bestemte posisjoner, noe som gjør disse modellene umulige. I dette arbeidet tar vi sikte på å løse dette problemet ved å etablere en alternativ metode for å utlede det negative settet fra det positive.
Vår metode er basert på det faktum at når funksjonene er vanlige mellom det negative og det positiv klasse, når modellen tar en beslutning, har den en tendens til å ignorere eller redusere avhengigheten av disse funksjonene (dvs. tildele lave vekter til disse funksjonene). I stedet blir modellen tvunget til å søke etter dypere og mindre åpenbare funksjoner. Deep learning-modeller lider generelt av langsom konvergens mens de trener på denne typen data. Denne metoden forbedrer imidlertid robustheten til modellen og sørger for generalisering. Vi rekonstruerer det negative settet som følger. Hver positive sekvens genererer en negativ sekvens. Den positive sekvensen er delt inn i 20 sekvenser. Deretter blir 12 undersøkelser valgt tilfeldig og erstattet tilfeldig. De resterende åtte påstandene er bevart. Denne prosessen er illustrert i figur 1. Bruk av denne prosessen på det positive settet resulterer i nye ikke-promotorsekvenser med konserverte deler fra promotersekvenser (de uendrede sekvensene, 8 sekvenser av 20). Disse parametrene muliggjør generering av et negativt sett som har 32 og 40% av sekvensene som inneholder konserverte deler av promotorsekvenser. Dette forholdet er funnet å være optimalt for å ha en robust promotor-prediktor som forklart i avsnitt 3.2.Fordi de konserverte delene inntar de samme posisjonene i de negative sekvensene, er de åpenbare motivene som TATA-box og TSS nå vanlige mellom de to settene med et forhold på 32 ~ 40%. Sekvenslogoene for de positive og negative settene for både human og mus TATA-promotordata er vist i henholdsvis figur 2, 3. Det kan sees at de positive og de negative settene deler de samme grunnmotivene på de samme posisjonene som TATA-motiv ved posisjon -30 og –25 bp og TSS ved posisjon +1 bp. Derfor er treningen mer utfordrende, men den resulterte modellen generaliserer godt.
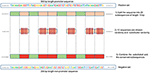
Figur 1. Illustrasjon av den negative settkonstruksjonsmetoden. Grønt representerer tilfeldig konserverte sekvenser mens rød representerer tilfeldig valgte og substituerte.

Figur 2. Sekvenslogoen i human TATA-promoter for både positivt sett (A) og negativt sett (B). Plottene viser bevaringen av funksjonelle motiver mellom de to settene.

Figur 3. Sekvenslogoen i mus TATA-promoter for både positivt sett (A) og negativt sett (B). Plottene viser bevaring av funksjonelle motiver mellom de to settene.
2.3. De foreslåtte modellene
Vi foreslår en dyp læringsmodell som kombinerer viklingslag med tilbakevendende lag som vist i figur 4. Den aksepterer en enkelt rå genomisk sekvens, S = {N1, N2,…, Nl} hvor N ∈ {A, C, G, T} og l er lengden på inngangssekvensen, som input og output en virkelig verdsatt score. Inngangen er en-hot-kodet og representert som en endimensjonal vektor med fire kanaler. Lengden på vektoren l = 300 og de fire kanalene er A, C, G og T og representert som (1 0 0 0), (0 1 0 0), (0 0 1 0), (0 0 0 1 ), henholdsvis. For å velge den modellen som har best resultat, har vi brukt rutenettmetode for å velge de beste hyperparametrene. Vi har prøvd forskjellige arkitekturer som CNN alene, LSTM alene, BiLSTM alene, CNN kombinert med LSTM. De innstilte hyperparametrene er antall sammenløpslag, kjernestørrelse, antall filtre i hvert lag, størrelsen på det maksimale samlingslaget, sannsynlighet for frafall og enhetene til Bi-LSTM-laget.
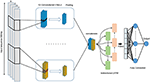
Figur 4. Arkitekturen til den foreslåtte DeePromoter-modellen.
Den foreslåtte modellen starter med flere konvolusjonslag som er justert parallelt og hjelper til med å lære de viktige motivene til inngangssekvensene med forskjellig vindusstørrelse. Vi bruker tre konvolusjonslag for ikke-TATA-promoter med vindusstørrelser på 27, 14 og 7, og to konvolusjonslag for TATA-promotorer med vindusstørrelser på 27, 14. Alle konvolusjonslag følges av ReLU-aktiveringsfunksjon (Glorot et al. , 2011), et maksimalt poolinglag med en vindusstørrelse på 6, og et dropout-lag med en sannsynlighet 0,5. Deretter blir utdataene fra disse lagene sammenkoblet og matet inn i et toveis langt korttidsminne (BiLSTM) (Schuster og Paliwal, 1997) med 32 noder for å fange avhengighetene mellom de lærte motivene fra konvolusjonslagene. De lærte funksjonene etter BiLSTM blir flatt og etterfulgt av frafall med en sannsynlighet på 0,5. Deretter legger vi til to fullt sammenkoblede lag for klassifisering. Den første har 128 noder og etterfulgt av ReLU og frafall med en sannsynlighet på 0,5 mens det andre laget brukes til prediksjon med en node- og sigmoid-aktiveringsfunksjon. BiLSTM lar informasjonen vedvare og lære langsiktige avhengigheter av sekvensielle prøver som DNA og RNA. Dette oppnås gjennom LSTM-strukturen som består av en minnecelle og tre porter som kalles inngangs-, utgangs- og glemmeporter. Disse portene er ansvarlige for å regulere informasjonen i minnecellen. I tillegg øker bruken av LSTM-modulen nettverksdybden mens antallet nødvendige parametere forblir lavt. Å ha et dypere nettverk muliggjør utvinning av mer komplekse funksjoner, og dette er hovedmålet med modellene våre, ettersom det negative settet inneholder harde prøver.
Keras-rammeverket brukes til å konstruere og trene de foreslåtte modellene (Chollet F. et. al., 2015). Adam optimizer (Kingma og Ba, 2014) brukes til å oppdatere parametrene med en læringsrate på 0,001. Batchstørrelsen er satt til 32 og antall epoker er satt til 50. Tidlig stopping brukes basert på valideringstap.
Resultater og diskusjon
3.1. Ytelsestiltak
I dette arbeidet bruker vi de allment aksepterte evalueringsmålingene for å evaluere ytelsen til de foreslåtte modellene.Disse beregningene er presisjon, tilbakekalling og Matthew korrelasjonskoeffisient (MCC), og de er definert som følger:
Hvor TP er sant positiv og representerer riktig identifiserte promotorsekvenser, er TN sant negativ og representerer riktig avviste promotersekvenser, FP er falsk positiv og representerer feil identifisert promotorsekvenser, og FN er falsk negativ og representerer feil avviste promotorsekvenser.
3.2. Effekt av det negative settet
Når vi analyserte de tidligere publiserte verkene for identifikasjon av promotorsekvenser, la vi merke til at ytelsen til disse verkene i stor grad avhenger av måten å forberede det negative datasettet på. De presterte veldig bra på datasettene de har utarbeidet, men de har et høyt falskt positivt forhold når de blir evaluert på et mer utfordrende datasett som inkluderer ikke-prompter sekvenser som har vanlige motiver med promotorsekvenser. For eksempel, i tilfelle av TATA-promoterdatasettet, vil de tilfeldig genererte sekvensene ikke ha TATA-motiv ved posisjon -30 og –25 bp, som igjen gjør klassifiseringsoppgaven lettere. Med andre ord var klassifisereren avhengig av tilstedeværelsen av TATA-motiv for å identifisere promotorsekvensen, og som et resultat var det lett å oppnå høy ytelse på datasettene de har utarbeidet. Modellene deres mislyktes imidlertid dramatisk når de hadde å gjøre med negative sekvenser som inneholdt TATA-motiv (harde eksempler). Presisjonen falt da den falske positive frekvensen økte. De klassifiserte ganske enkelt disse sekvensene som positive promotorsekvenser. En lignende analyse er gyldig for de andre promotormotivene. Derfor er hovedformålet med arbeidet vårt ikke bare å oppnå høy ytelse på et bestemt datasett, men også å forbedre modellens evne til å generalisere godt ved å trene på et utfordrende datasett.
For å illustrere dette poenget mer, trener vi og test vår modell på menneskelige og mus TATA promoter datasett med forskjellige metoder for forberedelse av negative sett. Det første eksperimentet ble utført ved å bruke tilfeldige utvalgte negative sekvenser fra ikke-kodende regioner i genomet (dvs. ligner den tilnærmingen som ble brukt i de forrige verkene). Bemerkelsesverdig oppnår vår foreslåtte modell nesten perfekt spådomsnøyaktighet (presisjon = 99%, tilbakekalling = 99%, Mcc = 98%) og (presisjon = 99%, tilbakekalling = 98%, Mcc = 97%) for henholdsvis menneske og mus . Disse høye resultatene forventes, men spørsmålet er om denne modellen kan opprettholde den samme ytelsen når den vurderes på et datasett som har harde eksempler. Svaret, basert på å analysere tidligere modeller, er nei. Det andre eksperimentet er utført ved hjelp av vår foreslåtte metode for å utarbeide datasettet som forklart i avsnitt 2.2. Vi forbereder de negative settene som inneholder konservert TATA-boks med forskjellige prosenter som 12, 20, 32 og 40%, og målet er å redusere gapet mellom presisjon og tilbakekalling. Dette sikrer at modellen vår lærer mer komplekse funksjoner i stedet for å lære bare tilstedeværelsen eller fraværet av TATA-box. Som vist i figurene 5A, B stabiliserer modellen seg i forholdet 32 ~ 40% for både TATA-promotors datasett for mennesker og mus.
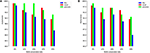
Figur 5. Effekten av forskjellige konserveringsforhold for TATA-motiv i negativt sett på ytelsen i tilfelle TATA-promotordatasett for både human (A) og mus (B) .
3.3. Resultater og sammenligning
I løpet av de siste årene har det blitt foreslått mange prediksjonsverktøy (Hutchinson, 1996; Scherf et al., 2000; Reese, 2001; Umarov og Solovyev, 2017). Noen av disse verktøyene er imidlertid ikke tilgjengelig for testing, og noen av dem krever mer informasjon i tillegg til de rå genomiske sekvensene. I denne studien sammenligner vi ytelsen til våre foreslåtte modeller med dagens toppmoderne verk, CNNProm, som ble foreslått av Umarov og Solovyev (2017) som vist i tabell 2. Generelt sett er de foreslåtte modellene, DeePromoter, klart bedre enn CNNProm i alle datasett med alle evalueringsberegninger. Mer spesifikt forbedrer DeePromoter presisjonen, tilbakekallingen og MCC i tilfelle av humant TATA-datasett med henholdsvis 0,18, 0,04 og 0,26. Når det gjelder humant ikke-TATA-datasett, forbedrer DeePromoter presisjonen med 0,39, tilbakekallingen med 0,12 og MCC med 0,66. Tilsvarende forbedrer DeePromoter presisjonen og MCC i tilfelle av mus TATA datasett med henholdsvis 0,24 og 0,31. I tilfelle av musesett som ikke er TATA, forbedrer DeePromoter presisjonen med 0,37, tilbakekallingen med 0,04 og MCC med 0,65. Disse resultatene bekrefter at CNNProm ikke klarer å avvise negative sekvenser med TATA-promoter, derfor har den høy falsk positiv. På den annen side er modellene våre i stand til å håndtere disse tilfellene mer vellykket, og falsk positiv rate er lavere sammenlignet med CNNProm.

Tabell 2. Sammenligning av DeePromoter med tilstanden til -the-art-metoden.
For videre analyser studerer vi effekten av alternerende nukleotider i hver posisjon på utgangsscore. Vi fokuserer på regionen –40 og 10 bp da den er vert for den viktigste delen av promotorsekvensen. For hver promotorsekvens i testsettet utfører vi beregningsmutasjonsskanning for å evaluere effekten av å mutere hver base av inngangssekvensen (150 substitusjoner på intervallet –40 ~ 10 bp-sekvens). Dette er illustrert i figur 6, 7 for henholdsvis humane og musedata-datasett. Blå farge representerer et fall i utgangsscore på grunn av mutasjon, mens den røde fargen representerer økningen av poengsummen på grunn av mutasjon. Vi merker at endring av nukleotidene til C eller G i regionen –30 og –25 bp reduserer utgangsscoren betydelig. Denne regionen er TATA-box, som er et veldig viktig funksjonelt motiv i promotorsekvensen. Dermed er modellen vår vellykket i stand til å finne viktigheten av denne regionen. I resten av posisjonene er C- og G-nukleotider mer å foretrekke enn A og T, spesielt når det gjelder musen. Dette kan forklares med det faktum at promotorregionen har flere C- og G-nukleotider enn A og T (Shi og Zhou, 2006).

Figur 6. Salinitetskartet over regionen –40 bp til 10 bp, som inkluderer TATA-boksen, i tilfelle humane TATA-promotorsekvenser.

Figur 7. Markeringskartet over regionen –40 bp til 10 bp, som inkluderer TATA-boksen, i tilfelle mus TATA-promotorsekvenser.
Konklusjon
Nøyaktig prediksjon av promotorsekvenser er viktig for å forstå den underliggende mekanismen i genreguleringsprosessen. I dette arbeidet utviklet vi DeePromoter -som er basert på en kombinasjon av nevrolusjonsnettverk og toveis LSTM- for å forutsi de korte eukaryote promotorsekvensene i tilfelle menneske og mus for både TATA og ikke-TATA-promoter. Den essensielle komponenten i dette arbeidet var å overvinne problemet med lav presisjon (høy falsk positiv hastighet) som ble lagt merke til i de tidligere utviklede verktøyene på grunn av avhengigheten av noen åpenbare funksjoner / motiver i sekvensen når man klassifiserte promoter- og ikke-promotorsekvenser. I dette arbeidet var vi spesielt interessert i å konstruere et hardt negativt sett som driver modellene mot å utforske sekvensen for dype og relevante funksjoner i stedet for bare å skille promoter- og ikke-promotersekvenser basert på eksistensen av noen funksjonelle motiver. De viktigste fordelene ved å bruke DeePromoter er at det reduserer antall falske positive spådommer betydelig, samtidig som det oppnås høy nøyaktighet på utfordrende datasett. DeePromoter overgikk den forrige metoden, ikke bare i ytelsen, men også i å overvinne problemet med høye falske positive spådommer. Det er anslått at dette rammeverket kan være nyttig i narkotikarelaterte applikasjoner og akademia.
Forfatterbidrag
MO og ZL utarbeidet datasettet, oppfattet algoritmen og gjennomførte eksperimentet og analyse. MO og HT forberedte webserveren og skrev manuskriptet med støtte fra ZL og KC. Alle forfattere diskuterte resultatene og bidro til det endelige manuskriptet.
Finansiering
Denne forskningen ble støttet av hjerneforskningsprogrammet til National Research Foundation (NRF) finansiert av den koreanske regjeringen ( MSIT) (nr. NRF-2017M3C7A1044815).
Erklæring om interessekonflikt
Forfatterne erklærer at forskningen ble utført i fravær av kommersielle eller økonomiske forhold som kunne tolkes som en potensiell interessekonflikt.
Bharanikumar, R., Premkumar, KAR og Palaniappan, A. (2018). Promoterforutsigelse: sekvensbasert modellering av escherichia coli σ70 promoterstyrke gir logaritmisk avhengighet mellom promotorstyrke og sekvens. PeerJ 6: e5862. doi: 10.7717 / peerj.5862
PubMed Abstract | CrossRef Fulltekst | Google Scholar
Glorot, X., Bordes, A. og Bengio, Y. (2011). «Deep sparse rectifier neurale nettverk,» i Proceedings of the Fourtenth International Conference on Artificial Intelligence and Statistics, (Fort Lauderdale, FL:) 315–323.
Google Scholar
Hutchinson, G. (1996). Forutsigelse av virveldyrpromotorregioner ved bruk av differensiell heksamerfrekvensanalyse. Bioinformatikk 12, 391–398.
PubMed Abstract | Google Scholar
Kingma, DP og Ba, J. (2014). Adam: en metode for stokastisk optimalisering. arXiv preprint arXiv: 1412.6980.
Google Scholar
Knudsen, S. (1999). Promoter2. 0: for anerkjennelse av polii-promotorsekvenser. Bioinformatics 15, 356–361.
PubMed Abstract | Google Scholar
Ponger, L. og Mouchiroud, D. (2002). Cpgprod: identifisering av cpg-øyene assosiert med transkripsjonsstartsteder i store genomiske pattedyrsekvenser. Bioinformatikk 18, 631–633. doi: 10.1093 / bioinformatics / 18.4.631
PubMed Abstract | CrossRef Fulltekst | Google Scholar
Quang, D. og Xie, X. (2016). Danq: et hybrid konvolusjonelt og tilbakevendende dypt nevralt nettverk for å kvantifisere funksjonen til dna-sekvenser. Nucleic Acids Res. 44, e107 – e107. doi: 10.1093 / nar / gkw226
PubMed Abstract | CrossRef Fulltekst | Google Scholar
Umarov, R. K. og Solovyev, V. V. (2017). Anerkjennelse av prokaryote og eukaryote promotorer ved hjelp av konvolusjonelle dyplærende nevrale nettverk. PLoS ONE 12: e0171410. doi: 10.1371 / journal.pone.0171410
PubMed Abstract | CrossRef Fulltekst | Google Scholar