Frontiers in Genetics
Inleiding
Promotors zijn de belangrijkste elementen die behoren tot niet-coderende regio’s in het genoom. Ze controleren grotendeels de activering of onderdrukking van de genen. Ze bevinden zich dichtbij en stroomopwaarts van de transcriptiestartplaats (TSS) van het gen. Het promotor-flankerende gebied van een gen kan veel cruciale korte DNA-elementen en -motieven bevatten (5 en 15 basen lang) die dienen als herkenningsplaatsen voor de eiwitten die zorgen voor juiste initiatie en regulatie van transcriptie van het stroomafwaartse gen (Juven-Gershon et al., 2008). De initiatie van gentranscriptie is de meest fundamentele stap in de regulering van genexpressie. De promotorkern is een minimale hoeveelheid DNA-sequentie die TSS conationeert en voldoende is om de transcriptie direct te starten. De lengte van de kernpromotor ligt typisch tussen 60 en 120 basenparen (bp).
De TATA-box is een promotor-subsequentie die aan andere moleculen aangeeft waar de transcriptie begint. Het werd “TATA-box” genoemd omdat de sequentie wordt gekenmerkt door herhalende T- en A-basenparen (TATAAA) (Baker et al., 2003). De overgrote meerderheid van de onderzoeken naar de TATA-box is uitgevoerd op mensen, gist, en Drosophila-genomen, maar vergelijkbare elementen zijn gevonden in andere soorten, zoals archaea en oude eukaryoten (Smale en Kadonaga, 2003). Bij mensen heeft 24% van de genen promotorregio’s die TATA-box bevatten (Yang et al., 2007 In eukaryoten bevindt de TATA-box zich op ~ 25 bp stroomopwaarts van de TSS (Xu et al., 2016). Het is in staat om de richting van transcriptie te bepalen en geeft ook de DNA-streng aan die moet worden gelezen. Eiwitten worden transcriptiefactoren genoemd binden aan verschillende niet-coderende regio’s, waaronder TATA-box en rekruteren een enzym genaamd RNA-polymerase, dat RNA synthetiseert uit DNA.
Vanwege de belangrijke rol van de promoters bij gentranscriptie, wordt nauwkeurige voorspelling van promotorplaatsen een vereiste stap in genexpressie, interpretatie van patronen en opbouw en begrip de functionaliteit van genetische regulerende netwerken. Er waren verschillende biologische experimenten voor identificatie van promotors, zoals mutatieanalyse (Matsumine et al., 1998) en immunoprecipitatietesten (Kim et al., 2004; Dahl en Collas, 2008). Deze methoden waren echter zowel duur als tijdrovend. Onlangs, met de ontwikkeling van de volgende generatie sequencing (NGS) (Behjati en Tarpey, 2013), is de sequentie van meer genen van verschillende organismen bepaald en zijn hun genelementen computationeel onderzocht (Zhang et al., 2011). Aan de andere kant heeft de innovatie van NGS-technologie geleid tot een dramatische daling van de kosten van de sequentiebepaling van het hele genoom, waardoor er meer sequentiegegevens beschikbaar zijn. De beschikbaarheid van gegevens trekt onderzoekers aan om rekenmodellen te ontwikkelen voor de voorspellingstaak van de promotor. Het is echter nog steeds een onvolledige taak en er is geen efficiënte software die promotors nauwkeurig kan voorspellen.
Promotor-voorspellers kunnen op basis van de gebruikte benadering worden onderverdeeld in drie groepen, namelijk een signaalgebaseerde benadering, en een inhoudgebaseerde benadering. , en de op GpG gebaseerde benadering. Signaalgebaseerde voorspellers richten zich op promotorelementen die verband houden met de RNA-polymerasebindingsplaats en negeren de niet-elementgedeelten van de sequentie. Als resultaat was de voorspellingsnauwkeurigheid zwak en niet bevredigend. Voorbeelden van signaalgebaseerde voorspellers zijn onder meer: PromoterScan (Prestridge, 1995) die de geëxtraheerde kenmerken van de TATA-box en een gewogen matrix van transcriptiefactorbindingsplaatsen met een lineaire discriminator gebruikte om promotorsequenties te classificeren als niet-promotorsequenties; Promoter2.0 (Knudsen, 1999) die de kenmerken uit verschillende boxen zoals TATA-Box, CAAT-Box en GC-Box haalde en ze voor classificatie doorgaf aan kunstmatige neurale netwerken (ANN); NNPP2.1 (Reese, 2001) die initiatorelement (Inr) en TATA-Box gebruikten voor het extraheren van kenmerken en een neuraal netwerk met tijdvertraging voor classificatie, en Down en Hubbard (2002) die TATA-Box gebruikten en een relevantievectormachine gebruikten (RVM) als classificator. Op inhoud gebaseerde voorspellers vertrouwden op het tellen van de frequentie van k-mer door een venster met k-lengte door de reeks te laten lopen. Deze methoden negeren echter de ruimtelijke informatie van de basenparen in de sequenties. Voorbeelden van op inhoud gebaseerde voorspellers zijn: PromFind (Hutchinson, 1996) dat de k-mer-frequentie gebruikte om de voorspelling van de hexameerpromotor uit te voeren; PromoterInspector (Scherf et al., 2000) die de regio’s identificeerde die promoters bevatten op basis van een gemeenschappelijke genomische context van polymerase II-promoters door te scannen op specifieke kenmerken die zijn gedefinieerd als motieven met variabele lengte; MCPromoter1.1 (Ohler et al., 1999) die een enkele geïnterpoleerde Markov-keten (IMC) van 5e orde gebruikte om promotersequenties te voorspellen.Ten slotte gebruikten op GpG gebaseerde voorspellers de locatie van GpG-eilanden als het promotorgebied of het eerste exongebied in de menselijke genen bevat gewoonlijk GpG-eilanden (Ioshikhes en Zhang, 2000; Davuluri et al., 2001; Lander et al., 2001; Ponger en Mouchiroud, 2002). Echter, slechts 60% van de promotors bevat GpG-eilanden, daarom is de voorspellingsnauwkeurigheid van dit soort voorspellers nooit meer dan 60% geweest.
Recent zijn sequentie-gebaseerde benaderingen gebruikt voor promoter-voorspelling. Yang et al. (2017) gebruikten verschillende kenmerk-extractiestrategieën om de meest relevante sequentie-informatie vast te leggen om enhancer-promotor-interacties te voorspellen. Lin et al. (2017) stelde een sequentie-gebaseerde voorspeller voor, genaamd “iPro70-PseZNC”, voor identificatie van de sigma70-promotor in de prokaryoot. Evenzo, Bharanikumar et al. (2018) stelde PromoterPredict voor om de sterkte van Escherichia coli-promoters te voorspellen op basis van een dynamische meervoudige regressiebenadering waarbij de sequenties werden weergegeven als positiegewichtmatrices (PWM). Kanhere en Bansal (2005) gebruikten de verschillen in stabiliteit van de DNA-sequentie tussen de promoter- en niet-promotersequenties om ze te onderscheiden. Xiao et al. (2018) introduceerde een twee-lagen-voorspeller genaamd iPSW (2L) -PseKNC voor identificatie van promotersequenties en voor de sterkte van de promoters door hybride kenmerken uit de sequenties te extraheren.
Alle bovengenoemde voorspellers vereisen domein- kennis om de functies met de hand te maken. Aan de andere kant maken op deep learning gebaseerde benaderingen het mogelijk om efficiëntere modellen te bouwen met behulp van onbewerkte gegevens (DNA / RNA-sequenties) rechtstreeks. Diep convolutioneel neuraal netwerk behaalde state-of-the-art resultaten in uitdagende taken zoals het verwerken van beeld, video, audio en spraak (Krizhevsky et al., 2012; LeCun et al., 2015; Schmidhuber, 2015; Szegedy et al. , 2015). Bovendien werd het met succes toegepast bij biologische problemen zoals DeepBind (Alipanahi et al., 2015), DeepCpG (Angermueller et al., 2017), branchepuntselectie (Nazari et al., 2018), voorspelling van alternatieve splitsingsplaatsen (Oubounyt et al., 2018), voorspelling van 2 “-Omethyleringsplaatsen (Tahir et al., 2018), kwantificering van de DNA-sequentie (Quang en Xie, 2016), subcellulaire lokalisatie van menselijk eiwit (Wei et al., 2018), enz. CNN kreeg onlangs veel aandacht in de promotorherkenningstaak. Zeer recent introduceerden Umarov en Solovyev (2017) CNNprom voor het discrimineren van korte promotersequenties, deze op CNN gebaseerde architectuur behaalde hoge resultaten bij het classificeren van promoter- en niet-promotersequenties. Daarna werd dit model verbeterd door Qian et al. (2018), waar de auteurs de SVM-classificator (Support Vector Machine) gebruikten om de belangrijkste elementen van de promotorsequentie te inspecteren. Vervolgens werden de meest invloedrijke elementen ongecomprimeerd gehouden terwijl de minder belangrijke werden gecomprimeerd. Dit proces resulteerde in betere prestaties. Onlangs werd een identificatiemodel voor lange promoters voorgesteld door Umarov et al. (2019) waarin de auteurs zich concentreerden op de identificatie van de TSS-positie.
In alle bovengenoemde werken werd de negatieve set geëxtraheerd uit niet-promotorregio’s van het genoom. Wetende dat de promotersequenties uitsluitend rijk zijn aan specifieke functionele elementen zoals TATA-box die zich op –30 ~ –25 bp bevindt, GC-Box die zich bevindt op –110 ~ -80 bp, CAAT-Box die zich bevindt op – 80 ~ -70 bp, enz. Dit resulteert in een hoge classificatienauwkeurigheid vanwege de enorme ongelijkheid tussen de positieve en negatieve monsters in termen van sequentiestructuur. Bovendien wordt de classificatietaak moeiteloos te bereiken, de CNN-modellen zullen bijvoorbeeld gewoon vertrouwen op de aan- of afwezigheid van sommige motieven op hun specifieke posities om de beslissing over het sequentietype te nemen. Deze modellen hebben dus een zeer lage precisie / gevoeligheid (hoog fout-positief) wanneer ze worden getest op genomische sequenties die promotormotieven hebben, maar het zijn geen promotersequenties. Het is bekend dat er meer TATAAA-motieven in het genoom zijn dan degene die tot de promotorregio’s behoren. Alleen al de DNA-sequentie van het menselijke chromosoom 1, ftp://ftp.ensembl.org/pub/release-57/fasta/homo_sapiens/dna/, bevat 151 656 TATAAA-motieven. Het is meer dan het geschatte maximale aantal genen in het totale menselijke genoom. Ter illustratie van dit probleem merken we op dat bij het testen van deze modellen op niet-promotersequenties die een TATA-box hebben, ze de meeste van deze sequenties verkeerd classificeren. Daarom, om een robuuste classificator te genereren, moet de negatieve set zorgvuldig worden geselecteerd, aangezien deze de kenmerken bepaalt die door de classificator zullen worden gebruikt om de klassen te onderscheiden. Het belang van dit idee is aangetoond in eerdere werken zoals (Wei et al., 2014). In dit werk pakken we dit probleem voornamelijk aan en stellen een benadering voor die enkele van de positieve klasse functionele motieven in de negatieve klasse integreert om de afhankelijkheid van het model van deze motieven te verminderen.We gebruiken een CNN in combinatie met LSTM-model om sequentiekenmerken van menselijke en muis TATA en niet-TATA eukaryote promotors te analyseren en computationele modellen te bouwen die nauwkeurig korte promotersequenties kunnen onderscheiden van niet-promotersequenties.
Materialen en methoden
2.1. Dataset
De datasets, die worden gebruikt voor het trainen en testen van de voorgestelde promotorvoorspeller, worden verzameld van mens en muis. Ze bevatten twee onderscheidende klassen van de promotors, namelijk TATA-promoters (d.w.z. de sequenties die TATA-box bevatten) en niet-TATA-promoters. Deze datasets zijn opgebouwd uit Eukaryotic Promoter Database (EPDnew) (Dreos et al., 2012). De EPDnew is een nieuwe sectie onder de bekende EPD-dataset (Périer et al., 2000) die een niet-redundante verzameling van eukaryote POL II-promoters bevat waarvan de startplaats voor transcriptie experimenteel is bepaald. Het biedt promotors van hoge kwaliteit in vergelijking met de ENSEMBL-promotercollectie (Dreos et al., 2012) en het is publiekelijk toegankelijk op https://epd.epfl.ch//index.php. We hebben TATA- en niet-TATA-promoter genomische sequenties voor elk organisme gedownload van EPDnew. Deze operatie resulteerde in het verkrijgen van vier datasets van de promotor, namelijk: Human-TATA, Human-non-TATA, Mouse-TATA en Mouse-non-TATA. Voor elk van deze datasets wordt een negatieve set (niet-promotersequenties) met dezelfde grootte als de positieve geconstrueerd op basis van de voorgestelde aanpak zoals beschreven in de volgende sectie. De details over het aantal promotorsequenties voor elk organisme worden gegeven in Tabel 1. Alle sequenties hebben een lengte van 300 bp en werden geëxtraheerd van -249 ~ + 50 bp (+1 verwijst naar TSS-positie). Als kwaliteitscontrole hebben we 5-voudige kruisvalidatie gebruikt om het voorgestelde model te beoordelen. In dit geval worden 3-vouwen gebruikt voor training, 1-vouw wordt gebruikt voor validatie en de resterende vouw wordt gebruikt voor testen. Het voorgestelde model wordt dus 5 keer getraind en de algehele prestatie van het 5-voudige wordt berekend.

Tabel 1. Statistieken van de vier datasets die in dit onderzoek zijn gebruikt.
2.2. Constructie van negatieve datasets
Om een model te trainen dat de classificatie van promoter- en niet-promotersequenties nauwkeurig kan uitvoeren, moeten we de negatieve set (niet-promotersequenties) zorgvuldig kiezen. Dit punt is cruciaal bij het maken van een model dat in staat is om goed te generaliseren, en daarom zijn precisie kan behouden wanneer het wordt geëvalueerd op meer uitdagende datasets. Eerdere werken, zoals (Qian et al., 2018), construeerden een negatieve set door willekeurig fragmenten te selecteren uit genoom-niet-promotorregio’s. Het is duidelijk dat deze benadering niet helemaal redelijk is, want als er geen kruising is tussen positieve en negatieve sets. Het model zal dus gemakkelijk basisfuncties vinden om de twee klassen te scheiden. Het TATA-motief kan bijvoorbeeld in alle positieve sequenties op een specifieke positie worden gevonden (normaal 28 bp stroomopwaarts van de TSS, tussen –30 en –25 pb in onze dataset). Daarom zal het willekeurig maken van een negatieve set die dit motief niet bevat, hoge prestaties leveren in deze dataset. Het model slaagt er echter niet in om negatieve sequenties te classificeren die TATA-motief als promotors hebben. Kort gezegd is de grootste fout in deze benadering dat bij het trainen van een deep learning-model het alleen leert om de positieve en negatieve klassen te onderscheiden op basis van de aan- of afwezigheid van enkele eenvoudige kenmerken op specifieke posities, wat deze modellen onpraktisch maakt. In dit werk proberen we dit probleem op te lossen door een alternatieve methode te vinden om de negatieve reeks af te leiden van de positieve.
Onze methode is gebaseerd op het feit dat wanneer de kenmerken gemeenschappelijk zijn tussen de negatieve en de positieve klasse, het model heeft de neiging om bij het nemen van de beslissing zijn afhankelijkheid van deze kenmerken te negeren of te verminderen (dwz lage gewichten aan deze kenmerken toe te kennen). In plaats daarvan wordt het model gedwongen te zoeken naar diepere en minder voor de hand liggende kenmerken. Deep learning-modellen hebben doorgaans te lijden onder een langzame convergentie tijdens het trainen op dit soort gegevens. Deze methode verbetert echter de robuustheid van het model en zorgt voor generalisatie. We reconstrueren de negatieve verzameling als volgt. Elke positieve reeks genereert een negatieve reeks. De positieve reeks is verdeeld in 20 subreeksen. Vervolgens worden 12 subreeksen willekeurig gekozen en willekeurig vervangen. De overige 8 subreeksen zijn behouden. Dit proces wordt geïllustreerd in figuur 1. Het toepassen van dit proces op de positieve set resulteert in nieuwe niet-promotorsequenties met geconserveerde delen van promotorsequenties (de ongewijzigde subsequenties, 8 subsequenties van de 20). Deze parameters maken het mogelijk om een negatieve set te genereren die 32 en 40% van zijn sequenties bevat die geconserveerde gedeelten van promotersequenties bevatten. Deze verhouding blijkt optimaal te zijn voor het hebben van een robuuste promotorvoorspeller, zoals uitgelegd in paragraaf 3.2.Omdat de geconserveerde delen dezelfde posities innemen in de negatieve sequenties, zijn de voor de hand liggende motieven zoals TATA-box en TSS nu gemeenschappelijk tussen de twee sets met een verhouding van 32 ~ 40%. De sequentielogo’s van de positieve en negatieve sets voor zowel de gegevens van de TATA-promotor van zowel de mens als de muis worden weergegeven in respectievelijk figuren 2 en 3. Het is te zien dat de positieve en de negatieve sets dezelfde basismotieven delen op dezelfde posities, zoals het TATA-motief op de positie -30 en –25 bp en de TSS op de positie +1 bp. Daarom is de training uitdagender, maar het resulterende model genereert goed.
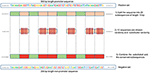
Figuur 1. Illustratie van de bouwwijze van de negatieve set. Groen vertegenwoordigt de willekeurig geconserveerde subreeksen, terwijl rood de willekeurig gekozen en vervangen subreeksen vertegenwoordigt.

Figuur 2. Het sequentielogo in menselijke TATA-promoter voor zowel positieve set (A) als negatieve set (B). De grafieken tonen het behoud van de functionele motieven tussen de twee sets.

Figuur 3. Het sequentielogo in de TATA-promoter van de muis voor zowel positieve set (A) als negatieve set (B). De grafieken tonen het behoud van de functionele motieven tussen de twee sets.
2.3. De voorgestelde modellen
We stellen een diep leermodel voor dat convolutielagen combineert met terugkerende lagen, zoals weergegeven in figuur 4. Het accepteert een enkele ruwe genomische sequentie, S = {N1, N2,…, Nl} waarbij N ∈ {A, C, G, T} en l is de lengte van de invoerreeks, als invoer en uitvoer een score met reële waarde. De invoer is one-hot gecodeerd en wordt weergegeven als een eendimensionale vector met vier kanalen. De lengte van de vector l = 300 en de vier kanalen zijn A, C, G en T en worden weergegeven als (1 0 0 0), (0 1 0 0), (0 0 1 0), (0 0 0 1 ), respectievelijk. Om het best presterende model te selecteren, hebben we een rasterzoekmethode gebruikt om de beste hyperparameters te kiezen. We hebben verschillende architecturen geprobeerd, zoals alleen CNN, alleen LSTM, alleen BiLSTM, CNN gecombineerd met LSTM. De afgestemde hyperparameters zijn het aantal convolutielagen, de grootte van de kernel, het aantal filters in elke laag, de grootte van de maximale poolinglaag, de uitvalkans en de eenheden van de Bi-LSTM-laag.
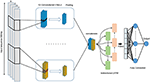
Figuur 4. De architectuur van het voorgestelde DeePromoter-model.
Het voorgestelde model begint met meerdere convolutielagen die parallel zijn uitgelijnd en helpen bij het leren van de belangrijke motieven van de invoersequenties met verschillende venstergroottes. We gebruiken drie convolutielagen voor niet-TATA-promoters met venstergroottes van 27, 14 en 7, en twee convolutielagen voor TATA-promoters met venstergroottes van 27, 14. Alle convolutielagen worden gevolgd door de ReLU-activeringsfunctie (Glorot et al. , 2011), een max pooling-laag met een venstergrootte van 6 en een dropout-laag met een kans van 0,5. Vervolgens worden de outputs van deze lagen samengevoegd en ingevoerd in een bidirectioneel langetermijngeheugen (BiLSTM) (Schuster en Paliwal, 1997) laag met 32 knooppunten om de afhankelijkheden tussen de aangeleerde motieven van de convolutielagen vast te leggen. De geleerde functies na BiLSTM worden afgeplat en gevolgd door uitval met een waarschijnlijkheid van 0,5. Vervolgens voegen we twee volledig verbonden lagen toe voor classificatie. De eerste heeft 128 knooppunten en wordt gevolgd door ReLU en drop-out met een waarschijnlijkheid van 0,5, terwijl de tweede laag wordt gebruikt voor voorspelling met één knooppunt en sigmoïde activeringsfunctie. BiLSTM zorgt ervoor dat de informatie blijft bestaan en leert de afhankelijkheden op lange termijn van opeenvolgende monsters zoals DNA en RNA. Dit wordt bereikt door de LSTM-structuur die is samengesteld uit een geheugencel en drie poorten genaamd input, output en vergeet-poorten. Deze poorten zijn verantwoordelijk voor het regelen van de informatie in de geheugencel. Bovendien vergroot het gebruik van de LSTM-module de netwerkdiepte terwijl het aantal vereiste parameters laag blijft. Het hebben van een dieper netwerk maakt het mogelijk om meer complexe functies te extraheren en dit is het hoofddoel van onze modellen, aangezien de negatieve set harde monsters bevat.
Het Keras-raamwerk wordt gebruikt voor het construeren en trainen van de voorgestelde modellen (Chollet F. et al., 2015). Adam optimizer (Kingma en Ba, 2014) wordt gebruikt voor het updaten van de parameters met een leertempo van 0,001. De batchgrootte is ingesteld op 32 en het aantal tijdvakken is ingesteld op 50. Vroegtijdig stoppen wordt toegepast op basis van validatieverlies.
Resultaten en bespreking
3.1. Prestatiemaatstaven
In dit werk gebruiken we de algemeen aanvaarde evaluatiestatistieken om de prestaties van de voorgestelde modellen te evalueren.Deze statistieken zijn precisie, herinnering en Matthew-correlatiecoëfficiënt (MCC), en ze worden als volgt gedefinieerd:
Waar TP echt positief is en correct geïdentificeerde promotorsequenties vertegenwoordigt, is TN echt negatief en vertegenwoordigt correct afgewezen promotorsequenties, FP is vals positief en vertegenwoordigt onjuist geïdentificeerd promotersequenties en FN is vals-negatief en staat voor ten onrechte afgewezen promotersequenties.
3.2. Effect van de negatieve set
Bij het analyseren van de eerder gepubliceerde werken voor identificatie van promotersequenties hebben we gemerkt dat de prestatie van die werken sterk afhangt van de manier waarop de negatieve dataset wordt voorbereid. Ze presteerden erg goed op de datasets die ze hebben opgesteld, maar ze hebben een hoge fout-positieve ratio wanneer ze worden geëvalueerd op een meer uitdagende dataset die niet-prompter-sequenties bevat met gemeenschappelijke motieven met promotersequenties. In het geval van de TATA-promotor-dataset zullen de willekeurig gegenereerde sequenties bijvoorbeeld geen TATA-motief hebben op de posities -30 en –25 bp, wat op zijn beurt de taak van classificatie gemakkelijker maakt. Met andere woorden, hun classificator was afhankelijk van de aanwezigheid van het TATA-motief om de promotersequentie te identificeren en als resultaat was het gemakkelijk om hoge prestaties te behalen met de datasets die ze hadden voorbereid. Hun modellen faalden echter dramatisch bij het omgaan met negatieve sequenties die TATA-motief bevatten (harde voorbeelden). De precisie nam af naarmate het percentage vals-positieven toenam. Ze classificeerden deze sequenties eenvoudig als positieve promotorsequenties. Een gelijkaardige analyse is geldig voor de andere promotormotieven. Daarom is het belangrijkste doel van ons werk niet alleen het bereiken van hoge prestaties op een specifieke dataset, maar ook het verbeteren van het vermogen van het model om goed te generaliseren door te trainen op een uitdagende dataset.
Om dit punt beter te illustreren, trainen we en test ons model op de datasets van de TATA-promotor van mens en muis met verschillende methoden voor de voorbereiding van negatieve sets. Het eerste experiment wordt uitgevoerd met behulp van willekeurig bemonsterde negatieve sequenties uit niet-coderende gebieden van het genoom (d.w.z. vergelijkbaar met de benadering die in de vorige werken werd gebruikt). Opmerkelijk is dat ons voorgestelde model een bijna perfecte voorspellingsnauwkeurigheid bereikt (precisie = 99%, terugroepen = 99%, Mcc = 98%) en (precisie = 99%, terugroepen = 98%, Mcc = 97%) voor respectievelijk mens en muis. . Deze hoge resultaten worden verwacht, maar de vraag is of dit model dezelfde prestaties kan behouden als het wordt geëvalueerd op een dataset met harde voorbeelden. Het antwoord, gebaseerd op het analyseren van de eerdere modellen, is nee. Het tweede experiment wordt uitgevoerd met behulp van onze voorgestelde methode voor het voorbereiden van de dataset, zoals uitgelegd in paragraaf 2.2. We bereiden de negatiefsets voor die een geconserveerde TATA-box bevatten met verschillende percentages zoals 12, 20, 32 en 40% en het doel is om de kloof tussen de precisie en de terugroepactie te verkleinen. Dit zorgt ervoor dat ons model meer complexe functies leert in plaats van alleen de aan- of afwezigheid van TATA-box te leren. Zoals getoond in figuren 5A, B, stabiliseert het model op de verhouding 32 ~ 40% voor zowel menselijke als muis TATA-promoter-datasets.
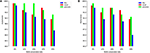
Figuur 5. Het effect van verschillende conserveringsverhoudingen van TATA-motief in de negatieve set op de prestaties in het geval van een TATA-promotor-dataset voor zowel mens (A) als muis (B) .
3.3. Resultaten en vergelijking
In de afgelopen jaren zijn tal van voorspellingsinstrumenten voor de promotorregio voorgesteld (Hutchinson, 1996; Scherf et al., 2000; Reese, 2001; Umarov en Solovyev, 2017). Sommige van deze tools zijn echter niet publiekelijk beschikbaar om te testen en sommige vereisen meer informatie naast de ruwe genomische sequenties. In deze studie vergelijken we de prestaties van onze voorgestelde modellen met het huidige state-of-the-art werk, CNNProm, dat werd voorgesteld door Umarov en Solovyev (2017) zoals weergegeven in tabel 2. Over het algemeen zijn de voorgestelde modellen, DeePromoter, presteert duidelijk beter dan CNNProm in alle datasets met alle evaluatiestatistieken. Meer specifiek verbetert DeePromoter de precisie, terugroepactie en MCC in het geval van menselijke TATA-dataset met respectievelijk 0,18, 0,04 en 0,26. In het geval van menselijke niet-TATA-dataset verbetert DeePromoter de precisie met 0,39, de terugroepactie met 0,12 en MCC met 0,66. Evenzo verbetert DeePromoter de precisie, en MCC in het geval van muis TATA-gegevensset met respectievelijk 0,24 en 0,31. In het geval van een niet-TATA-dataset van muizen verbetert DeePromoter de precisie met 0,37, de terugroepactie met 0,04 en MCC met 0,65. Deze resultaten bevestigen dat CNNProm er niet in slaagt om negatieve sequenties met TATA-promotor te verwerpen, daarom heeft het een hoog vals-positief resultaat. Aan de andere kant zijn onze modellen in staat om deze gevallen met meer succes aan te pakken en is het percentage valse positieven lager in vergelijking met CNNProm.

Tabel 2. Vergelijking van de DeePromoter met de status van -the-art methode.
Voor verdere analyses bestuderen we het effect van alternerende nucleotiden op elke positie op de outputscore. We concentreren ons op de regio -40 en 10 bp omdat het het belangrijkste deel van de promotersequentie herbergt. Voor elke promotorsequentie in de testset voeren we computationele mutatiescanning uit om het effect te evalueren van het muteren van elke base van de invoersubsequentie (150 substituties op de interval -40 ~ 10 bp subsequentie). Dit wordt geïllustreerd in de figuren 6, 7 voor respectievelijk menselijke en muis-TATA-datasets. Blauwe kleur vertegenwoordigt een daling van de outputscore als gevolg van mutatie, terwijl de rode kleur de toename van de score vertegenwoordigt als gevolg van mutatie. We merken dat het veranderen van de nucleotiden naar C of G in de regio –30 en –25 bp de outputscore significant verlaagt. Dit gebied is de TATA-box, wat een zeer belangrijk functioneel motief is in de promotersequentie. Zo weet ons model met succes het belang van deze regio te achterhalen. In de rest van de posities hebben C- en G-nucleotiden meer de voorkeur dan A en T, vooral in het geval van de muis. Dit kan worden verklaard door het feit dat het promotorgebied meer C- en G-nucleotiden heeft dan A en T (Shi en Zhou, 2006).

Figuur 6. De saliency-kaart van de regio -40 bp tot 10 bp, die de TATA-box bevat, in het geval van menselijke TATA-promotersequenties.

Figuur 7. De saliency-kaart van de regio -40 bp tot 10 bp, die de TATA-box bevat, in het geval van muis TATA-promotersequenties.
Conclusie
Nauwkeurige voorspelling van promotersequenties is essentieel om het onderliggende mechanisme van het genregulatieproces te begrijpen. In dit werk hebben we DeePromoter ontwikkeld -die is gebaseerd op een combinatie van convolutie neuraal netwerk en bidirectionele LSTM- om de korte eukaryote promotersequenties te voorspellen in het geval van mens en muis voor zowel TATA- als niet-TATA-promoter. De essentiële component van dit werk was het overwinnen van het probleem van lage precisie (hoog percentage vals-positieven) dat werd opgemerkt in de eerder ontwikkelde tools vanwege de afhankelijkheid van een aantal duidelijke kenmerken / motieven in de sequentie bij het classificeren van promotor- en niet-promotorsequenties. In dit werk waren we vooral geïnteresseerd in het construeren van een harde negatieve set die de modellen ertoe aanzet de sequentie te onderzoeken op diepe en relevante kenmerken in plaats van alleen onderscheid te maken tussen de promoter- en niet-promotersequenties op basis van het bestaan van enkele functionele motieven. De belangrijkste voordelen van het gebruik van DeePromoter zijn dat het het aantal fout-positieve voorspellingen aanzienlijk vermindert en tegelijkertijd een hoge nauwkeurigheid bereikt bij uitdagende datasets. DeePromoter presteerde beter dan de vorige methode, niet alleen in de prestaties, maar ook in het overwinnen van het probleem van hoge fout-positieve voorspellingen. Verwacht wordt dat dit raamwerk nuttig kan zijn in drugsgerelateerde toepassingen en de academische wereld.
Bijdragen van auteurs
MO en ZL hebben de dataset voorbereid, het algoritme bedacht en het experiment uitgevoerd en analyse. MO en HT hebben de webserver voorbereid en het manuscript geschreven met steun van ZL en KC. Alle auteurs bespraken de resultaten en droegen bij aan het uiteindelijke manuscript.
Financiering
Dit onderzoek werd ondersteund door het Brain Research Program van de National Research Foundation (NRF), gefinancierd door de Koreaanse overheid ( MSIT) (nr. NRF-2017M3C7A1044815).
Verklaring over belangenconflicten
De auteurs verklaren dat het onderzoek is uitgevoerd bij afwezigheid van commerciële of financiële relaties die kunnen worden opgevat als een mogelijk belangenconflict.
Bharanikumar, R., Premkumar, KAR, en Palaniappan, A. (2018). Promoterpredict: sequentie-gebaseerde modellering van escherichia coli σ70 promotorsterkte levert een logaritmische afhankelijkheid op tussen promotorsterkte en sequentie. PeerJ 6: e5862. doi: 10.7717 / peerj.5862
PubMed Abstract | CrossRef Volledige tekst | Google Scholar
Glorot, X., Bordes, A., en Bengio, Y. (2011). “Deep sparse rectifier neurale netwerken”, in Proceedings of the Fourteenth International Conference on Artificial Intelligence and Statistics, (Fort Lauderdale, FL 🙂 315–323.
Google Scholar
Hutchinson, G. (1996). De voorspelling van promotorregio’s van gewervelde dieren met behulp van differentiële hexameerfrequentieanalyse. Bioinformatics 12, 391–398.
PubMed Abstract | Google Scholar
Kingma, DP en Ba, J. (2014). Adam: een methode voor stochastische optimalisatie. arXiv preprint arXiv: 1412.6980.
Google Scholar
Knudsen, S. (1999). Promoter2.0: voor de herkenning van polii-promotersequenties. Bioinformatics 15, 356-361.
PubMed Abstract | Google Scholar
Ponger, L. en Mouchiroud, D. (2002). Cpgprod: identificatie van cpg-eilanden die zijn geassocieerd met startlocaties voor transcriptie in grote genomische zoogdiersequenties. Bioinformatics 18, 631-633. doi: 10.1093 / bioinformatics / 18.4.631
PubMed Abstract | CrossRef Volledige tekst | Google Scholar
Quang, D. en Xie, X. (2016). Danq: een hybride convolutioneel en recidiverend diep neuraal netwerk voor het kwantificeren van de functie van dna-sequenties. Nucleic Acids Res. 44, e107-e107. doi: 10.1093 / nar / gkw226
PubMed Abstract | CrossRef Volledige tekst | Google Scholar
Umarov, R. K. en Solovyev, V. V. (2017). Erkenning van prokaryote en eukaryote promotors met behulp van convolutionele neurale netwerken voor diep leren. PLoS ONE 12: e0171410. doi: 10.1371 / journal.pone.0171410
PubMed Abstract | CrossRef Volledige tekst | Google Scholar