Frontiers in Genetics (Polski)
Wprowadzenie
Promotory to kluczowe elementy należące do niekodujących regionów w genomie. W dużej mierze kontrolują aktywację lub represję genów. Znajdują się one w pobliżu i powyżej miejsca startu transkrypcji genu (TSS). Region flankujący promotora genu może zawierać wiele kluczowych krótkich elementów DNA i motywów (długości 5 i 15 zasad), które służą jako miejsca rozpoznawania białek, które zapewniają właściwa inicjacja i regulacja transkrypcji dolnego genu (Juven-Gershon et al., 2008). Inicjacja transkryptu genów jest najbardziej podstawowym krokiem w regulacji ekspresji genów. Rdzeń promotora to minimalny odcinek sekwencji DNA, który łączy TSS i wystarczający do bezpośredniej inicjacji transkrypcji. Długość promotora rdzeniowego zazwyczaj mieści się w zakresie od 60 do 120 par zasad (bp).
TATA-box jest podsekwencją promotora, która wskazuje innym cząsteczkom, gdzie zaczyna się transkrypcja. Został nazwany „TATA-box”, ponieważ jego sekwencja charakteryzuje się powtarzającymi się parami zasad T i A (TATAAA) (Baker i wsp., 2003). Zdecydowana większość badań nad pudełkiem TATA została przeprowadzona na ludziach, drożdżach, i Drosophila, jednakże podobne elementy znaleziono u innych gatunków, takich jak archeony i starożytne eukarioty (Smale i Kadonaga, 2003). W przypadku człowieka 24% genów ma regiony promotorowe zawierające TATA-box (Yang i in., 2007 U eukariotów TATA-box znajduje się około 25 pz przed TSS (Xu i wsp., 2016). Jest w stanie określić kierunek transkrypcji, a także wskazuje nić DNA do odczytania. Białka zwane czynnikami transkrypcyjnymi wiążą się z kilkoma niekodującymi regionami, w tym TATA-box i rekrutują enzym zwany polimerazą RNA, który syntetyzuje RNA z DNA.
Ze względu na ważną rolę promotorów w transkrypcji genów, dokładne przewidywanie miejsc promotorowych staje się wymagany krok w ekspresji genów, interpretacji wzorców oraz budowaniu i zrozumieniu funkcjonalność genetycznych sieci regulacyjnych. Przeprowadzono różne eksperymenty biologiczne w celu identyfikacji promotorów, takie jak analiza mutacji (Matsumine i wsp., 1998) i testy immunoprecypitacji (Kim i wsp., 2004; Dahl i Collas, 2008). Jednak metody te były zarówno drogie, jak i czasochłonne. Niedawno, wraz z rozwojem sekwencjonowania nowej generacji (NGS) (Behjati i Tarpey, 2013), zsekwencjonowano więcej genów różnych organizmów, a ich elementy genowe zbadano obliczeniowo (Zhang et al., 2011). Z drugiej strony innowacja technologii NGS spowodowała dramatyczny spadek kosztów sekwencjonowania całego genomu, a zatem dostępnych jest więcej danych dotyczących sekwencjonowania. Dostępność danych przyciąga naukowców do opracowania modeli obliczeniowych do zadania przewidywania promotora. Jest to jednak wciąż niekompletne zadanie i nie ma wydajnego oprogramowania, które mogłoby dokładnie przewidzieć promotorów.
Predyktory promotorów można podzielić na trzy grupy w oparciu o zastosowane podejście, a mianowicie podejście oparte na sygnale, podejście oparte na treści i podejście oparte na GpG. Predyktory oparte na sygnale koncentrują się na elementach promotorowych związanych z miejscem wiązania polimerazy RNA i ignorują nieelementowe części sekwencji. W rezultacie dokładność przewidywania była słaba i niezadowalająca. Przykłady predyktorów opartych na sygnale obejmują: PromoterScan (Prestridge, 1995), który wykorzystywał wyekstrahowane cechy TATA-box i ważoną macierz miejsc wiązania czynników transkrypcyjnych z liniowym dyskryminatorem w celu klasyfikacji sekwencji promotorowych od niepromotorowych; Promoter2.0 (Knudsen, 1999), który wyodrębnił cechy z różnych modułów, takich jak TATA-Box, CAAT-Box i GC-Box, i przekazał je do sztucznych sieci neuronowych (ANN) w celu klasyfikacji; NNPP2.1 (Reese, 2001), który wykorzystywał element inicjujący (Inr) i TATA-Box do ekstrakcji cech i opóźnioną w czasie sieć neuronową do klasyfikacji oraz Down i Hubbard (2002), który wykorzystywał TATA-Box i maszyny wektorów trafności (RVM) jako klasyfikator. Predyktory oparte na treści polegały na zliczaniu częstotliwości k-mer przez uruchomienie okna długości k w sekwencji. Jednak metody te ignorują informacje przestrzenne o parach zasad w sekwencjach. Przykłady predyktorów opartych na treści obejmują: PromFind (Hutchinson, 1996), który wykorzystał częstotliwość k-mer do wykonania przewidywania promotora heksamerowego; PromoterInspector (Scherf et al., 2000), który zidentyfikował regiony zawierające promotory w oparciu o wspólny kontekst genomowy promotorów polimerazy II poprzez skanowanie w poszukiwaniu specyficznych cech określonych jako motywy o zmiennej długości; MCPromoter1.1 (Ohler i wsp., 1999), w którym zastosowano pojedynczy interpolowany łańcuch Markowa (IMC) piątego rzędu do przewidywania sekwencji promotorowych.Wreszcie, predyktory oparte na GpG wykorzystywały lokalizację wysp GpG jako region promotora lub pierwszy region egzonu w ludzkich genach zwykle zawiera wyspy GpG (Ioshikhes i Zhang, 2000; Davuluri i in., 2001; Lander i in., 2001; Ponger i Mouchiroud, 2002). Jednak tylko 60% promotorów zawiera wyspy GpG, dlatego dokładność przewidywania tego rodzaju predyktorów nigdy nie przekroczyła 60%.
Ostatnio do przewidywania promotora wykorzystano podejścia oparte na sekwencjach. Yang i in. (2017) wykorzystali różne strategie ekstrakcji cech, aby uchwycić najbardziej istotne informacje o sekwencji w celu przewidzenia interakcji wzmacniacz-promotor. Lin i in. (2017) zaproponowali predyktor oparty na sekwencji, nazwany „iPro70-PseZNC”, do identyfikacji promotora sigma70 u prokariota. Podobnie Bharanikumar i in. (2018) zaproponowali PromoterPredict, aby przewidzieć siłę promotorów Escherichia coli w oparciu o dynamiczne podejście regresji wielokrotnej, w którym sekwencje były reprezentowane jako macierze wagi pozycji (PWM). Kanhere i Bansal (2005) wykorzystali różnice w stabilności sekwencji DNA między sekwencjami promotorowymi i nie-promotorowymi w celu ich rozróżnienia. Xiao i in. (2018) wprowadzili dwuwarstwowy predyktor o nazwie iPSW (2L) -PseKNC do identyfikacji sekwencji promotorowych, a także siłę promotorów poprzez wyodrębnianie cech hybrydowych z sekwencji.
Wszystkie wyżej wymienione predyktory wymagają domeny- wiedzę w celu ręcznego wykonania funkcji. Z drugiej strony podejścia oparte na głębokim uczeniu się umożliwiają budowanie bardziej wydajnych modeli bezpośrednio przy użyciu surowych danych (sekwencji DNA / RNA). Głębokie konwolucyjne sieci neuronowe osiągnęły najnowocześniejsze wyniki w trudnych zadaniach, takich jak przetwarzanie obrazu, wideo, audio i mowy (Krizhevsky i in., 2012; LeCun i in., 2015; Schmidhuber, 2015; Szegedy i in. , 2015). Ponadto został z powodzeniem zastosowany w problemach biologicznych, takich jak DeepBind (Alipanahi et al., 2015), DeepCpG (Angermueller et al., 2017), selekcja punktów rozgałęzienia (Nazari et al., 2018), przewidywanie alternatywnych miejsc splicingu (Oubounyt et al., 2018), przewidywanie miejsc 2 „-Ometylacji (Tahir et al., 2018), kwantyfikacja sekwencji DNA (Quang i Xie, 2016), lokalizacja subkomórkowa ludzkiego białka (Wei et al., 2018) itd. Ponadto, CNN ostatnio zyskało dużą uwagę w zadaniu rozpoznawania promotora. Niedawno Umarov i Solovyev (2017) wprowadzili CNNprom do rozróżniania krótkich sekwencji promotorowych, ta architektura oparta na CNN osiągnęła wysokie wyniki w klasyfikowaniu sekwencji promotora i bez promotora. Następnie model ten został ulepszony autorstwa Qian i wsp. (2018), w którym autorzy wykorzystali klasyfikator maszyny wektorów nośnych (SVM) do zbadania najważniejszych elementów sekwencji promotora, a następnie elementy o największym wpływie były nieskompresowane podczas kompresji mniej ważnych. Ten proces zaowocował lepszą wydajnością. Ostatnio, długi model identyfikacji promotora został zaproponowany przez Umarova i wsp. (2019), w którym autorzy skupili się na identyfikacji pozycji TSS.
We wszystkich wyżej wymienionych pracach zestaw negatywny został wyekstrahowany z niepromotorowych regionów genomu. Wiedząc, że sekwencje promotorowe są bogate wyłącznie w specyficzne elementy funkcjonalne, takie jak TATA-box, który znajduje się przy –30 ~ –25 pz, GC-Box, który znajduje się przy –110 ~ –80 pz, CAAT-Box, który znajduje się przy – 80 ~ 70 pz, itp. Skutkuje to wysoką dokładnością klasyfikacji ze względu na ogromne rozbieżności między próbkami dodatnimi i ujemnymi pod względem struktury sekwencji. Ponadto zadanie klasyfikacji staje się łatwe do osiągnięcia, na przykład modele CNN będą polegać po prostu na obecności lub braku niektórych motywów w ich określonych pozycjach, aby podjąć decyzję o typie sekwencji. Zatem modele te mają bardzo niską precyzję / czułość (wysoki wynik fałszywie dodatni), gdy są testowane na sekwencjach genomowych, które mają motywy promotorowe, ale nie są sekwencjami promotorowymi. Powszechnie wiadomo, że w genomie znajduje się więcej motywów TATAAA niż motywów należących do regionów promotorowych. Na przykład, sama sekwencja DNA ludzkiego chromosomu 1, ftp://ftp.ensembl.org/pub/release-57/fasta/homo_sapiens/dna/, zawiera 151 656 motywów TATAAA. To więcej niż przybliżona maksymalna liczba genów w całym ludzkim genomie. Aby zilustrować ten problem, zauważamy, że podczas testowania tych modeli na sekwencjach niepromotorowych, które mają kasetę TATA, błędnie klasyfikują większość z tych sekwencji. Dlatego w celu wygenerowania solidnego klasyfikatora należy starannie dobierać zestaw ujemny, ponieważ określa on cechy, które będą używane przez klasyfikator w celu rozróżnienia klas. Znaczenie tego pomysłu zostało wykazane w poprzednich pracach, takich jak (Wei i in., 2014). W tej pracy zajmujemy się głównie tym problemem i proponujemy podejście, które integruje niektóre z pozytywnych motywów funkcjonalnych klasy w klasie negatywnej, aby zmniejszyć zależność modelu od tych motywów.Wykorzystujemy CNN w połączeniu z modelem LSTM do analizy charakterystyk sekwencji ludzkich i mysich promotorów eukariotycznych TATA i innych niż TATA i budujemy modele obliczeniowe, które mogą dokładnie odróżniać krótkie sekwencje promotorów od tych bez promotorów.
Materiały i metody
2.1. Zbiór danych
Zbiory danych, które są używane do szkolenia i testowania proponowanego predyktora promotora, są zbierane od człowieka i myszy. Zawierają one dwie charakterystyczne klasy promotorów, mianowicie promotory TATA (tj. Sekwencje zawierające kasetę TATA) i promotory inne niż TATA. Te zbiory danych zostały zbudowane na podstawie bazy danych Eukariotic Promoter Database (EPDnew) (Dreos i in., 2012). EPDnew to nowa sekcja w ramach dobrze znanego zbioru danych EPD (Périer et al., 2000), który zawiera adnotacje jako zbiór eukariotycznych promotorów POL II, gdzie miejsce startu transkrypcji zostało określone eksperymentalnie. Zapewnia promotory wysokiej jakości w porównaniu z kolekcją promotorów ENSEMBL (Dreos i in., 2012) i jest publicznie dostępny pod adresem https://epd.epfl.ch//index.php. Pobraliśmy sekwencje genomowe promotora TATA i inne niż TATA dla każdego organizmu z EPDnew. W wyniku tej operacji uzyskano cztery zestawy danych promotora, a mianowicie: Human-TATA, Human-non-TATA, Mouse-TATA i Mouse-non-TATA. Dla każdego z tych zestawów danych, zestaw negatywny (sekwencje bez promotora) o tej samej wielkości co zestaw pozytywny jest konstruowany w oparciu o proponowane podejście, jak opisano w następnej sekcji. Szczegóły dotyczące liczby sekwencji promotorowych dla każdego organizmu podano w tabeli 1. Wszystkie sekwencje mają długość 300 pz i zostały wyekstrahowane od -249 ~ + 50 pz (+1 odnosi się do pozycji TSS). Jako kontrolę jakości zastosowaliśmy 5-krotną weryfikację krzyżową, aby ocenić proponowany model. W tym przypadku 3-krotne są używane do trenowania, 1-krotne do walidacji, a pozostałe krotności są używane do testowania. Dlatego proponowany model jest trenowany 5 razy i obliczana jest ogólna wydajność 5-krotnego.

Tabela 1. Statystyki czterech zbiorów danych wykorzystanych w tym badaniu.
2.2. Konstrukcja negatywnego zbioru danych
Aby wytrenować model, który może dokładnie przeprowadzić klasyfikację sekwencji promotora i bez promotora, musimy ostrożnie wybrać zestaw ujemny (sekwencje bez promotora). Ten punkt ma kluczowe znaczenie dla stworzenia modelu zdolnego do dobrego uogólniania, a tym samym zdolnego do utrzymania precyzji podczas oceny na bardziej wymagających zbiorach danych. Wcześniejsze prace, takie jak (Qian et al., 2018), konstruowały negatywny zestaw poprzez losową selekcję fragmentów z regionów genomu nie będących promotorami. Oczywiście takie podejście nie jest całkowicie rozsądne, ponieważ nie ma przecięcia między dodatnimi i ujemnymi zbiorami. W ten sposób model z łatwością znajdzie podstawowe cechy oddzielające dwie klasy. Na przykład, motyw TATA można znaleźć we wszystkich sekwencjach dodatnich w określonej pozycji (zwykle 28 pz przed TSS, między –30 a –25 pz w naszym zbiorze danych). Dlatego losowe utworzenie negatywnego zestawu, który nie zawiera tego motywu, zapewni wysoką wydajność w tym zbiorze danych. Jednak model zawodzi w klasyfikowaniu negatywnych sekwencji, które mają motyw TATA jako promotory. Krótko mówiąc, główną wadą tego podejścia jest to, że trenując model głębokiego uczenia, uczy się on jedynie rozróżniania klas pozytywnych i negatywnych na podstawie obecności lub braku pewnych prostych cech na określonych pozycjach, co sprawia, że modele te są niewykonalne. W tej pracy dążymy do rozwiązania tego problemu, ustanawiając alternatywną metodę wyprowadzenia zestawu negatywnego z pozytywnego.
Nasza metoda opiera się na fakcie, że ilekroć cechy są wspólne między tym, co negatywne i pozytywnej klasy model ma tendencję do pomijania lub ograniczania swojej zależności od tych cech, podejmując decyzję (tj. przypisuje im niskie wagi). Zamiast tego model jest zmuszony szukać głębszych i mniej oczywistych cech. Modele uczenia głębokiego generalnie cierpią na powolną konwergencję podczas uczenia na tego typu danych. Jednak ta metoda poprawia solidność modelu i zapewnia generalizację. Zbiór ujemny rekonstruujemy w następujący sposób. Każda pozytywna sekwencja generuje jedną negatywną sekwencję. Sekwencja pozytywna jest podzielona na 20 podsekwencji. Następnie losowo wybiera się 12 podciągów i losowo podstawia. Pozostałe 8 podciągów jest konserwowanych. Proces ten zilustrowano na Figurze 1. Zastosowanie tego procesu do zbioru pozytywnego daje w wyniku nowe sekwencje niepromotorowe z konserwatywnymi częściami z sekwencji promotorowych (niezmienione podsekwencje, 8 z 20). Te parametry umożliwiają generowanie negatywnego zestawu, który ma 32 i 40% jego sekwencji zawierających konserwatywne części sekwencji promotorowych. Stwierdzono, że stosunek ten jest optymalny do posiadania silnego predyktora promotora, jak wyjaśniono w sekcji 3.2.Ponieważ części konserwatywne zajmują te same pozycje w sekwencjach negatywnych, oczywiste motywy, takie jak TATA-box i TSS, są teraz wspólne między dwoma zestawami ze stosunkiem 32 ~ 40%. Loga sekwencji pozytywnych i negatywnych zestawów danych zarówno dla ludzkiego, jak i mysiego promotora TATA przedstawiono odpowiednio na Figurach 2, 3. Można zauważyć, że zestawy pozytywne i negatywne mają te same motywy podstawowe w tych samych pozycjach, takie jak motyw TATA w pozycji -30 i -25 pz oraz TSS w pozycji +1 pz. Dlatego szkolenie jest trudniejsze, ale wynikowy model dobrze uogólnia.
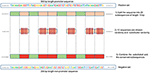
Rysunek 1. Ilustracja metody konstrukcji zestawu ujemnego. Zielony reprezentuje losowo zachowane podsekwencje, a czerwony reprezentuje losowo wybrane i podstawione.

Rysunek 2. Logo sekwencji w ludzkim promotorze TATA zarówno dla zestawu dodatniego (A), jak i zestawu ujemnego (B). Wykresy pokazują zachowanie motywów funkcjonalnych między dwoma zestawami.

Rysunek 3. Logo sekwencji w mysim promotorze TATA zarówno dla zestawu dodatniego (A), jak i zestawu ujemnego (B). Wykresy pokazują zachowanie motywów funkcjonalnych między dwoma zestawami.
2.3. Proponowane modele
Proponujemy model głębokiego uczenia, który łączy warstwy splotu z powtarzającymi się warstwami, jak pokazano na rysunku 4. Akceptuje pojedynczą nieprzetworzoną sekwencję genomową, S = {N1, N2,…, Nl} gdzie N ∈ {A, C, G, T} i l to długość sekwencji wejściowej, jako wejście i wyjście wyniku o wartości rzeczywistej. Dane wejściowe są zakodowane na gorąco i reprezentowane jako jednowymiarowy wektor z czterema kanałami. Długość wektora l = 300, a cztery kanały to A, C, G i T i reprezentowane jako (1 0 0 0), (0 1 0 0), (0 0 1 0), (0 0 0 1 ), odpowiednio. W celu wybrania najlepszego modelu wykorzystaliśmy metodę przeszukiwania siatki, aby wybrać najlepsze hiperparametry. Wypróbowaliśmy różne architektury, takie jak sam CNN, sam LSTM, sam BiLSTM, CNN w połączeniu z LSTM. Dostrojone hiperparametry to liczba warstw splotu, rozmiar jądra, liczba filtrów w każdej warstwie, rozmiar maksymalnej warstwy puli, prawdopodobieństwo rezygnacji i jednostki warstwy Bi-LSTM.
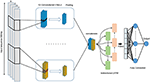
Rysunek 4. Architektura proponowanego modelu DeePromoter.
Proponowany model zaczyna się od wielu warstw splotów, które są wyrównane równolegle i pomagają w nauce ważnych motywów sekwencji wejściowych o różnej wielkości okna. Używamy trzech warstw splotu dla promotora innego niż TATA o rozmiarze okienka 27, 14 i 7 oraz dwóch warstw splotu dla promotorów TATA o rozmiarze okienka 27, 14. Po wszystkich warstwach splotu następuje funkcja aktywacji ReLU (Glorot et al. , 2011), maksymalna warstwa puli o rozmiarze okna 6 i warstwa odpadająca z prawdopodobieństwem 0,5. Następnie wyniki tych warstw są łączone ze sobą i wprowadzane do dwukierunkowej warstwy pamięci długoterminowej (BiLSTM) (Schuster i Paliwal, 1997) z 32 węzłami w celu uchwycenia zależności między wyuczonymi motywami z warstw splotu. Wyuczone cechy po BiLSTM są spłaszczane, a po nich następuje rezygnacja z prawdopodobieństwem 0,5. Następnie dodajemy dwie w pełni połączone warstwy do klasyfikacji. Pierwsza ma 128 węzłów, po której następuje ReLU i zanik z prawdopodobieństwem 0,5, podczas gdy druga warstwa jest używana do przewidywania z jednym węzłem i sigmoidalną funkcją aktywacji. BiLSTM umożliwia utrwalenie informacji i poznanie długoterminowych zależności między kolejnymi próbkami, takimi jak DNA i RNA. Osiąga się to poprzez strukturę LSTM, która składa się z komórki pamięci i trzech bram zwanych bramkami wejściowymi, wyjściowymi i zapomnienia. Te bramki są odpowiedzialne za regulowanie informacji w komórce pamięci. Ponadto wykorzystanie modułu LSTM zwiększa głębokość sieci, podczas gdy liczba wymaganych parametrów pozostaje niska. Posiadanie głębszej sieci umożliwia wyodrębnianie bardziej złożonych funkcji i jest to główny cel naszych modeli, ponieważ zbiór negatywów zawiera twarde próbki.
Framework Keras jest używany do konstruowania i uczenia proponowanych modeli (Chollet F. et al., 2015). Optymalizator Adama (Kingma i Ba, 2014) służy do aktualizacji parametrów z szybkością uczenia 0,001. Wielkość wsadu jest ustawiona na 32, a liczbę epok na 50. Wczesne zatrzymanie jest stosowane na podstawie utraty walidacji.
Wyniki i dyskusja
3.1. Miary wydajności
W tej pracy używamy szeroko przyjętych metryk oceny do oceny wydajności proponowanych modeli.Te metryki to precyzja, zapamiętanie i współczynnik korelacji Matthew (MCC). Są one zdefiniowane w następujący sposób:
Tam, gdzie TP jest prawdziwie dodatni i reprezentuje prawidłowo zidentyfikowane sekwencje promotorowe, TN jest prawdziwie ujemny i reprezentuje poprawnie odrzucone sekwencje promotora, FP jest fałszywie dodatni i reprezentuje nieprawidłowo zidentyfikowany sekwencje promotora, a FN jest fałszywie ujemna i reprezentuje nieprawidłowo odrzucone sekwencje promotorowe.
3.2. Efekt zbioru negatywnego
Analizując opublikowane wcześniej prace pod kątem identyfikacji sekwencji promotorowych zauważyliśmy, że wykonanie tych prac w dużej mierze zależy od sposobu przygotowania negatywnego zbioru danych. Działali bardzo dobrze na przygotowanych przez siebie zestawach danych, jednak mają wysoki współczynnik wyników fałszywie dodatnich, gdy są oceniani na bardziej wymagającym zestawie danych, który zawiera sekwencje inne niż prompter, mające wspólne motywy z sekwencjami promotorowymi. Na przykład, w przypadku zbioru danych promotora TATA, losowo generowane sekwencje nie będą miały motywu TATA na pozycji -30 i –25 pz, co z kolei ułatwia klasyfikację. Innymi słowy, ich klasyfikator zależał od obecności motywu TATA do identyfikacji sekwencji promotora, w wyniku czego łatwo było osiągnąć wysoką wydajność na przygotowanych przez nich zbiorach danych. Jednak ich modele zawiodły dramatycznie w przypadku negatywnych sekwencji zawierających motyw TATA (trudne przykłady). Precyzja spadła wraz ze wzrostem odsetka wyników fałszywie dodatnich. Po prostu sklasyfikowali te sekwencje jako pozytywne sekwencje promotorowe. Podobna analiza dotyczy innych motywów promotora. Dlatego głównym celem naszej pracy jest nie tylko osiągnięcie wysokiej wydajności na określonym zestawie danych, ale także zwiększenie zdolności modelu do dobrego uogólniania poprzez szkolenie na wymagającym zestawie danych.
Aby lepiej zilustrować ten punkt, trenujemy i przetestuj nasz model na zestawach danych ludzkiego i mysiego promotora TATA z różnymi metodami przygotowania negatywnych zestawów. Pierwszy eksperyment jest przeprowadzany przy użyciu losowo próbkowanych sekwencji negatywnych z niekodujących regionów genomu (tj. Podobnie do podejścia zastosowanego w poprzednich pracach). Co ciekawe, nasz proponowany model osiąga niemal doskonałą dokładność przewidywania (precyzja = 99%, przypominanie = 99%, Mcc = 98%) i (precyzja = 99%, przypominanie = 98%, Mcc = 97%) odpowiednio dla człowieka i myszy . Te wysokie wyniki są oczekiwane, ale pytanie brzmi, czy ten model może zachować taką samą wydajność, gdy jest oceniany na zestawie danych, który ma twarde przykłady. Odpowiedź, oparta na analizie poprzednich modeli, brzmi: nie. Drugie doświadczenie przeprowadza się za pomocą proponowanej przez nas metody przygotowania zbioru danych, jak wyjaśniono w sekcji 2.2. Przygotowujemy negatywne zestawy, które zawierają konserwowane TATA-box z różnymi wartościami procentowymi, takimi jak 12, 20, 32 i 40%, a celem jest zmniejszenie luki między precyzją a przypomnieniem. Gwarantuje to, że nasz model uczy się bardziej złożonych funkcji, zamiast uczyć się tylko obecności lub braku TATA-box. Jak pokazano na rysunkach 5A, B, model stabilizuje się w stosunku 32 ~ 40% dla zestawów danych promotora TATA zarówno ludzkich, jak i mysich.
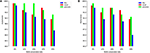
Rysunek 5. Wpływ różnych współczynników zachowania motywu TATA w zestawie negatywnym na wydajność w przypadku zbioru danych promotora TATA zarówno dla człowieka (A), jak i myszy (B) .
3.3. Wyniki i porównanie
W ostatnich latach zaproponowano wiele narzędzi do przewidywania regionów promotora (Hutchinson, 1996; Scherf i in., 2000; Reese, 2001; Umarov i Solovyev, 2017). Jednak niektóre z tych narzędzi nie są publicznie dostępne do testowania, a niektóre z nich wymagają więcej informacji poza surowymi sekwencjami genomowymi. W tym badaniu porównujemy wydajność proponowanych przez nas modeli z najnowszymi pracami CNNProm, które zaproponowali Umarow i Sołowjew (2017), jak pokazano w tabeli 2. Ogólnie rzecz biorąc, proponowane modele, DeePromoter, wyraźnie przewyższają CNNProm we wszystkich zestawach danych ze wszystkimi wskaźnikami oceny. Dokładniej, DeePromoter poprawia precyzję, rozpoznawanie i MCC w przypadku zbioru danych TATA człowieka o odpowiednio 0,18, 0,04 i 0,26. W przypadku zbioru danych innych niż TATA, DeePromoter poprawia precyzję o 0,39, przywołanie o 0,12, a MCC o 0,66. Podobnie DeePromoter poprawia precyzję, a MCC w przypadku mysiego zbioru danych TATA o odpowiednio 0,24 i 0,31. W przypadku zestawu danych myszy innych niż TATA, DeePromoter poprawia precyzję o 0,37, przywołanie o 0,04, a MCC o 0,65. Wyniki te potwierdzają, że CNNProm nie odrzuca negatywnych sekwencji z promotorem TATA, dlatego ma wysoki poziom fałszywie dodatni. Z drugiej strony nasze modele są w stanie lepiej radzić sobie z takimi przypadkami, a wskaźnik fałszywie pozytywnych wyników jest niższy w porównaniu z CNNProm.

Tabela 2. Porównanie DeePromoter ze stanem Metoda sztuki.
W celu dalszych analiz badamy wpływ przemiennych nukleotydów w każdej pozycji na wynik wyjściowy. Skupiamy się na regionie –40 i 10 pz, ponieważ zawiera on najważniejszą część sekwencji promotora. Dla każdej sekwencji promotora w zestawie testowym wykonujemy obliczeniowe skanowanie mutacji, aby ocenić efekt mutacji każdej zasady podsekwencji wejściowej (150 podstawień w przedziale -40 ~ 10 pz podsekwencji). Jest to zilustrowane na rysunkach 6, 7 odpowiednio dla zestawów danych TATA człowieka i myszy. Kolor niebieski oznacza spadek wyniku wynikowego z powodu mutacji, podczas gdy kolor czerwony reprezentuje wzrost wyniku spowodowany mutacją. Zauważamy, że zmiana nukleotydów na C lub G w regionie –30 i –25 pz znacznie zmniejsza wynik wyjściowy. Ten region to kaseta TATA, która jest bardzo ważnym motywem funkcjonalnym w sekwencji promotora. W ten sposób nasz model jest w stanie z powodzeniem określić znaczenie tego regionu. W pozostałych pozycjach nukleotydy C i G są bardziej korzystne niż A i T, zwłaszcza w przypadku myszy. Można to wytłumaczyć faktem, że region promotora ma więcej nukleotydów C i G niż A i T (Shi i Zhou, 2006).

Rysunek 6. Mapa istotności regionu od 40 pz do 10 pz, która obejmuje TATA-box, w przypadku ludzkich sekwencji promotorowych TATA.

Rysunek 7. Mapa istotności regionu od 40 pz do 10 pz, która zawiera kasetę TATA, w przypadku mysich sekwencji promotora TATA.
Wniosek
Dokładne przewidywanie sekwencji promotorów jest niezbędne do zrozumienia mechanizmu leżącego u podstaw procesu regulacji genów. W tej pracy opracowaliśmy DeePromoter – który jest oparty na połączeniu splotowej sieci neuronowej i dwukierunkowej LSTM – do przewidywania krótkich sekwencji promotora eukariotów w przypadku człowieka i myszy zarówno dla promotora TATA, jak i nie-TATA. Istotnym elementem tej pracy było przezwyciężenie problemu niskiej precyzji (wysoki odsetek wyników fałszywie dodatnich), zauważonego we wcześniej opracowanych narzędziach, ze względu na oparcie się na pewnych oczywistych cechach / motywach w sekwencji podczas klasyfikacji sekwencji promotorowych i niepromotorowych. W tej pracy byliśmy szczególnie zainteresowani skonstruowaniem twardego zestawu negatywnego, który kieruje modele w kierunku eksploracji sekwencji pod kątem głębokich i istotnych cech, zamiast rozróżniania tylko sekwencji promotora i niepromotorowych na podstawie istnienia pewnych motywów funkcjonalnych. Główną zaletą korzystania z DeePromoter jest to, że znacznie zmniejsza liczbę fałszywie pozytywnych przewidywań, jednocześnie osiągając wysoką dokładność w trudnych zestawach danych. DeePromoter przewyższył poprzednią metodę nie tylko pod względem wydajności, ale także w przezwyciężeniu problemu wysokich fałszywie pozytywnych prognoz. Przewiduje się, że ramy te mogą być pomocne w zastosowaniach związanych z lekami i w środowisku akademickim.
Wkład autorów
MO i ZL przygotowali zbiór danych, opracowali algorytm i przeprowadzili eksperyment oraz analiza. MO i HT przygotowali serwer WWW i napisali manuskrypt przy wsparciu ZL i KC. Wszyscy autorzy omówili wyniki i przyczynili się do powstania ostatecznej wersji manuskryptu.
Finansowanie
Badania te były wspierane przez Program Badań Mózgu National Research Foundation (NRF) finansowany przez rząd Korei ( MSIT) (nr NRF-2017M3C7A1044815).
Oświadczenie o konflikcie interesów
Autorzy oświadczają, że badanie zostało przeprowadzone przy braku jakichkolwiek powiązań handlowych lub finansowych, które można by zinterpretować jako potencjalny konflikt interesów.
Bharanikumar, R., Premkumar, KAR i Palaniappan, A. (2018). Promoterpredict: oparte na sekwencjach modelowanie siły promotora σ70 Escherichia coli daje logarytmiczną zależność między siłą promotora a sekwencją. PeerJ 6: e5862. doi: 10.7717 / peerj.5862
PubMed Abstract | Pełny tekst CrossRef | Google Scholar
Glorot, X., Bordes, A. i Bengio, Y. (2011). „Deep sparse rectifier neural networks”, w: Proceedings of the Fourteenth International Conference on Artificial Intelligence and Statistics, (Fort Lauderdale, FL:) 315–323.
Google Scholar
Hutchinson, G. (1996). Przewidywanie regionów promotora kręgowców przy użyciu analizy częstotliwości różnicowej heksameru. Bioinformatics 12, 391–398.
PubMed Abstract | Google Scholar
Kingma, DP i Ba, J. (2014). Adam: metoda optymalizacji stochastycznej. arXiv preprint arXiv: 1412.6980.
Google Scholar
Knudsen, S. (1999) .Promoter2. 0: for the rozpoznawanie sekwencji promotora polii, Bioinformatics 15, 356–361.
PubMed Abstract | Google Scholar
Ponger, L. i Mouchiroud, D. (2002). Cpgprod: identyfikacja wysp cpg związanych z miejscami startu transkrypcji w dużych genomowych sekwencjach ssaków. Bioinformatics 18, 631–633. doi: 10.1093 / bioinformatyka / 18.4.631
PubMed Abstract | Pełny tekst CrossRef | Google Scholar
Quang, D. and Xie, X. (2016). Danq: hybrydowa splotowa i rekurencyjna głęboka sieć neuronowa do ilościowego określania funkcji sekwencji DNA. Nucleic Acids Res. 44, e107 – e107. doi: 10.1093 / nar / gkw226
PubMed Abstract | Pełny tekst CrossRef | Google Scholar
Umarov, R. K. and Solovyev, V. V. (2017). Rozpoznawanie promotorów prokariotycznych i eukariotycznych za pomocą konwolucyjnych sieci neuronowych uczenia głębokiego. PLoS ONE 12: e0171410. doi: 10.1371 / journal.pone.0171410
PubMed Abstract | Pełny tekst CrossRef | Google Scholar