Fronteiras em imunologia
Introdução
A pré-eclâmpsia é uma doença específica da gravidez que afeta de 3 a 5% de todas as gestações (1, 2). Manifesta-se com hipertensão de início recente após 20 semanas de gestação e proteinúria. A placenta é central na patogênese da doença ao conectar a mãe ao feto (3). A interface materno-fetal é a zona onde o tecido geneticamente diferente de origem fetal se encontra com a circulação materna, o endotélio e o sistema imunológico. Freqüentemente chamada de doença das teorias, um insight geralmente aceito é que a pré-eclâmpsia afeta o endotélio materno, causando distúrbios na função do endotélio, o que leva à hipertensão e proteinúria (2). Em sua forma grave, a pré-eclâmpsia pode causar retardo do crescimento fetal, prematuridade e, para a mãe, pode causar falência de órgãos nos rins e no fígado, bem como eclâmpsia. Além disso, a pré-eclâmpsia pode ter consequências cardiovasculares adversas de longo prazo para a mãe e o recém-nascido (4). O desenvolvimento da placenta é considerado inadequado especificamente no processo de transformação da artéria espiral materna, onde as células trofoblásticas invadem o lado materno em decídua e transformam as artérias em condutos de baixa resistência, substituindo também o endotélio materno (5–7). Normalmente, essa remodelação das artérias uterinas está ausente ou incompleta, especialmente na forma grave de pré-eclâmpsia. A alta resistência nas artérias uterinas contraídas causa fluxo sanguíneo turbulento no espaço interviloso da placenta, causando estresse oxidativo e danos mecânicos às árvores vilosas da placenta. Os danos resultantes aumentam a liberação placentária de micropartículas e mediadores inflamatórios, resultando em ativação e disfunção endotelial generalizada (8, 9). Esses eventos sequenciais na interface materno-fetal levam à hipertensão materna e outros sintomas, conforme descrito acima.
A síndrome HELLP foi caracterizada pela primeira vez em 1982 por Weinstein como uma síndrome separada, frequentemente representando junto com a pré-eclâmpsia, mas também vista isoladamente (10, 11). A síndrome HELLP é caracterizada por hemólise, enzimas hepáticas elevadas e plaquetas baixas. Freqüentemente, requer observação do nível de cuidado intensivo e terapia sintomática. A patogênese da síndrome ainda está nas sombras. A síndrome HELLP compartilha características comuns com microangiopatias trombóticas (TMAs), como púrpura trombocitopênica trombótica (TTP) e síndrome hemolítico-urêmica (SHU). Os TMAs estão presentes em diversos grupos de doenças com características comuns de hemólise microangiopática, trombocitopenia e lesão orgânica resultante de microtrombos. Na TTP, um defeito genético de, ou como na grande maioria dos casos, autoanticorpos adquiridos contra uma desintegrina e metaloproteinase com motivo de trombospondina tipo 1 13 (ADAMTS13), a enzima que cliva o fator de von Willebrand ativado (vWF), causa a formação de ativou multímeros de vWF nas células endoteliais, levando a trombos plaquetários em pequenos vasos e hemólise. HUS típica é causada por infecção bacteriana produtora de toxina Shiga (especialmente Escherichia coli), enquanto HUS atípica (aHUS) se refere ao tipo de TMA, em que deficiências genéticas nos reguladores da via alternativa do sistema complemento estão subjacentes, que, na presença de um gatilho pode causar a doença clínica (12, 13). O tratamento aprovado pela FDA para SHUa é a infusão do antagonista C5 do complemento eculizumabe, que evita a formação do complexo de ataque à membrana (MAC) (Figura 1). Clinicamente, o HELLP compartilha os mesmos sintomas dos TMAs clássicos: hemólise, trombocitopenia e distúrbios de órgãos observados no fígado. Dependendo da classificação TMA usada, a síndrome HELLP é geralmente categorizada como parte de TMAs secundários ou adquiridos (Tabela 1).
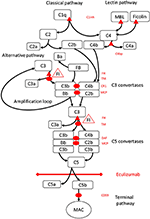
Figura 1. Eculizumab inibe a via terminal de ativação do complemento. A ativação do complemento pode ser iniciada por meio de três vias de ativação, todas levando à formação de C5 convertases que têm a capacidade de ativar a via terminal que leva à formação do MAC na superfície alvo. Esta ilustração esquemática do sistema complemento mostra seus ativadores mais relevantes (em fonte preta) e inibidores (em vermelho). Os ativadores podem ser ligados à membrana (oval) ou solúvel (triângulo). A via alternativa é ativada espontaneamente em todas as superfícies que não permitem a inibição pelo fator regulador solúvel H (FH). FH atua como o cofator para a inativação de C3b a iC3b pelo fator I (FI). A trombomodulina (TM) aumenta a atividade do cofator FH. O fator B clivado (FB) junto com o ativador C3b forma a convertase da via alternativa, que tem a capacidade de clivar C3 em C3b criando uma alça de amplificação de ativação da via alternativa.A via clássica de ativação do complemento pode ser iniciada, por exemplo, pela ligação de complexos imunes a C1q, enquanto a via da lectina é ativada por lectina de ligação de manoses (MBL) ou ligação de ficolinas a, por exemplo, padrões de carboidratos em micróbios. A ativação da via da lectina resulta em serina proteases 1 e 2 associadas à manose (MASP-1 e MASP-2, não ilustrado), clivando os componentes do complemento C4 e C2 para formar a via clássica convertase C4bC2b. O inibidor C1 (C1inh) e C4bp são os reguladores solúveis da via clássica, enquanto a proteína cofator de membrana (MCP) e o receptor do complemento 1 (CR1) são reguladores ligados à membrana das vias iniciais do complemento. A formação de C5 convertases inicia a via terminal de ativação do complemento e clivagem de C5 na ausência de reguladores ligados à superfície do fator de aceleração do decaimento (FAD) e MCP. A montagem do MAC é regulada pelo regulador de superfície ligada CD59 (protectina). A clivagem de C3 nas vias iniciais e C5 na via terminal libera anafilatoxinas C3a e C5a e resulta em inflamação. Eculizumab é um anticorpo recombinante humanizado contra a proteína C5 do complemento, que inibe a clivagem de C5 pelas convertases C5, regulando assim os efeitos pró-trombóticos e pró-inflamatórios da ativação do complemento. O paciente descrito neste relato de caso foi testado para mutações genéticas em genes que codificam para Fator H (CFH), FHR5 e MCP, CFI, CFB, THBD e ADAMTS13 (ADAMTS13), um regulador da via do FvW da cascata de coagulação (não mostrando). Os resultados do teste genético foram negativos.

Tabela 1. Critérios de diagnóstico HELLP e aHUS.
Neste estudo de caso, relatamos um paciente com TMA induzida pela gravidez complicada e o curso de tratamento bem-sucedido.
Apresentação do caso
Primigesta de 29 anos foi encaminhada com 34 + 2 anos de idade gestacional para o hospital da Universidade de Helsinque ambulatório com dor no estômago superior. Inicialmente, sua pressão arterial estava modestamente elevada (133/91 mmHg) e a vareta urinária positiva para proteína. A dor no estômago superior relatada inicialmente estava melhorando gradualmente. Na ultrassonografia, o feto apresentava perfil biofísico normal (BPP), a estimativa de peso estava na curva de crescimento de -2 DP. A cardiotocografia (CTG) era normal. A hemoglobina sanguínea (Hb) era 115 g / L, plaquetas 158 E9 / L (intervalo normal 150–360 E9 / L), alanina aminotransferase (ALT) era normal (23 U / L). A tira reagente urinária foi positiva para proteína (+2) e a proteinúria calculada foi de 1,6 g / 24 h. Decidiu-se iniciar o tratamento com cortisona para facilitar a maturação pulmonar do bebê. A paciente recebeu alta com plano de retorno no dia seguinte para check-up de controle e segunda dose de cortisona. Conforme programado, ela veio para o controle na semana gestacional 34 + 4. A pressão arterial era de 147/87 mmHg, ALT 23, plaquetas 177, CTG e o BPP do feto na ultrassonografia era normal. Ela teve alta e outro check-up foi agendado. Na tarde do mesmo dia, a dor no estômago superior voltou e piorou continuamente ao anoitecer. Ela voltou ao hospital às 2h20. Ela estava sentindo dores na parte superior do estômago, inquietação, e ela tinha vomitado duas vezes e estava sentindo tremor. A pressão arterial estava claramente elevada em 170/94 mmHg, a fita reagente de proteína na urina era fortemente positiva, a ALT estava elevada em 159, a Hb 122 e as plaquetas em 172. Ela foi internada na enfermaria de pré-natal. Às 4 da manhã ela estava com dor de cabeça. Foi iniciado medicamento anti-hipertensivo (Labetalol 100 mg três vezes). A excreção de proteínas na urina atingiu um pico noturno de 13 g / 24 h. Posteriormente, ela começou a vomitar, teve dor na parte superior do estômago, cefaleia e a monitorização CTG mostrou desaceleração. A paciente foi transferida às 7,11 da manhã para a enfermaria de parto e como o colo do útero estava com três centímetros de dilatação, as membranas fetais foram rompidas artificialmente para a indução do parto. Ao mesmo tempo, os testes de laboratório foram concluídos com Hb 122, plaquetas 172. A lactato desidrogenase (LD), no entanto, estava claramente elevada em 1231 U / L neste momento. No CTG, as desacelerações continuaram e com a continuação da bradicardia foi realizada cesariana de emergência. Lactente do sexo masculino (1960 g, −2 SD) nasceu às 7,25 da manhã com valor de pH da artéria umbilical de 7,05, BE −6,80, Apgar 1/6/8. A perda de sangue na operação foi de 400 ml.
Às 9h após a cesariana, as plaquetas da mãe estavam baixas, 49, com Hb de 102. À tarde, o ALT havia subido para 1800, LD 3570 , o nível de creatinina sérica era 153 (μmol / L), enquanto as plaquetas diminuíam para 33. Houve distúrbio na coagulação indicado por baixo nível de fibrinogênio (1,1 g / L, valores de referência 2–4 g / L) e alto nível de D -dímero de fibrina (30,2 mg / L, < 0,5 mg / L). Houve algum sangramento na ferida da cesariana, na qual suturas adicionais foram colocadas.Nesse momento, foram administradas oito unidades de plaquetas. O nível de potássio aumentou de 4,7 para 5,6 (mmol / L). Hemólise foi claramente observada. O teste de Coombs foi negativo. A excreção urinária foi de apenas 10 ml / h. O teste laboratorial mostrou claramente uma doença grave com sinais de danos nos rins e no fígado. Além disso, havia distúrbio no sistema de coagulação apresentando coagulação significativa e fibrinólise acentuada simultaneamente. A infusão de sulfato de magnésio foi iniciada por causa da hiperreflexia, considerada um sinal preditivo de convulsões, uma complicação grave da pré-eclâmpsia. Foi iniciada dexametasona 10 mg intravenosa e o paciente foi transferido para a unidade de terapia intensiva (UTI).
Além disso, exames laboratoriais foram emitidos para fins de diagnóstico diferencial de outras emergências médicas (Tabela 2). A atividade de ADAMTS13 era normal 62% (40-130%), o que exclui TTP. Os níveis de complemento sérico C3 (0,52 g / L, 0,71-1,41 g / L) e C4 (0,07 g / L, 0,12-0,34 g / L) estavam baixos. O nível de complexo terminal solúvel do complemento (C5b-9, 971 ng / mL, < 366 ng / mL) foi elevado no primeiro dia pós-parto. Não foram detectados anticorpos antifosfolípides, a sorologia para infecção para hepatites B e C e HIV negativa. A partir da amostra de fezes, os patógenos que causam SHU típico tiveram resultados negativos.

Tabela 2. Cronograma do diagnóstico e tratamento da doença.
A paciente foi tratada com tratamento de plasmaférese no primeiro e no segundo dia pós-parto e foi hemodializada três vezes ao longo do tratamento (dias 2, 4 e 6 pós-parto).
No terceiro dia pós-parto, a paciente estava estável e transferida de volta para a sala de recuperação do Hospital Feminino. e a terapia sintomática foi continuada. A hipertensão foi tratada com Amlodipina 10 mg duas vezes ao dia e Labetalol 200 mg três vezes ao dia. No quarto dia pós-parto, as plaquetas continuaram diminuindo e a paciente foi diagnosticada com SHUa. Freqüentemente o diagnóstico diferencial com síndrome HELLP A SHUa está na recuperação espontânea de pacientes HELLP, geralmente no terceiro dia pós-parto. O tratamento com eculizumabe foi iniciado (900 mg IV). Pati Ele recebeu vacina antipneumocócica e antibiótico profilático (penicilina). A paciente recebeu quatro doses semanais de eculizumab (900 mg) e começou a se recuperar rapidamente. Não necessitou de nova hemodiálise após a terceira hemodiálise no sexto dia pós-parto (Figura 2). Função renal corrigida gradualmente, contagem de plaquetas elevada e hemólise resolvida. Quatro semanas após o parto, os níveis plasmáticos de C3 e C4 foram normalizados.
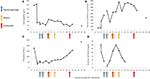
Figura 2. Valores laboratoriais selecionados observados durante os estágios iniciais da doença e o momento da troca plasmática, hemodiálise e administração de eculizumabe. No painel (A) está representado o desenvolvimento das medições de hemoglobina no sangue, no painel (B) o nível de creatinina sérica, no painel (C) o número de plaquetas e no painel (D) Dímero D dos valores de fibrina ao longo do 17 dia período de acompanhamento.
Em testes genéticos, nenhum polimorfismo de gene conhecido foi identificado. Ela foi testada para mutações em reguladores de complemento Fator H (CFH), Fator H relacionado à proteína 5 (FHR5) e proteína cofator de membrana (MCP), complemento do fator de inativação da via alternativa I (CFI), fator de ativação da via alternativa B (CFB ), e os seguintes componentes da cascata de coagulação: ADAMTS13, trombomodulina (THBD) e uma enzima intracelular, diacilglicerol quinase E (DGKE), cujas mutações são uma causa conhecida de SHUa (14). Além disso, anticorpos contra o fator H também não foram detectados. Nenhuma deficiência de C4 foi detectada.
Em resumo, nossa paciente tinha pré-eclâmpsia grave e preenchia os critérios diagnósticos para a síndrome HELLP. Embora o teste genético para SHUa tenha permanecido negativo, o curso clínico da doença (especialmente, lesão renal aguda grave) e a resposta ao tratamento (especialmente, eculizumabe) sugeriu que nossa paciente tinha SHUa induzida pela gravidez.
Discussão
A primigesta descrita acima foi diagnosticada com pré-eclâmpsia grave, síndrome HELLP e SHUa induzida pela gravidez. Nenhuma mutação genética conhecida, que poderia predispor a SHUa, foi descoberta. O diagnóstico diferencial entre os tipos de TMA, TTP, HUS / aHUS e TMAs secundários como a síndrome HELLP é importante. Especialmente a TTP deve ser identificada no início, já que a doença é tratada com troca de plasma diária rápida até a remissão (15).
Atualmente, os métodos de análise laboratorial para testar mutações genéticas que potencialmente causam SHUa são capazes de mostrar mutações em apenas até 40–60% dos casos de SHUa, deixando a possibilidade de casos falso-negativos. Portanto, um teste negativo para mutações não exclui um verdadeiro SHUa (16).Raramente a SHUa foi induzida após a gravidez e o parto. Nestes casos, durante os três anos seguintes ~ 50% desenvolveram doença renal crônica (DRC), alguns até mesmo doença renal em estágio terminal (ESKD). Quando mutações genéticas foram observadas, até 85% podem desenvolver CKD ou ESKD (17).
É geralmente aceito que em distúrbios hipertensivos da gravidez, a inflamação placentária resulta em disfunção endotelial. Se a integridade do endotélio for perturbada, isso resulta em ativação do complemento e coagulação (18). Também foi hipotetizado que o endotélio materno interrompido contribui para a morbidade materna tardia associada à pré-eclâmpsia grave e outros distúrbios hipertensivos da gravidez (2, 19). Foi demonstrado que o endotélio danificado funciona de maneira anormal mesmo anos após o desaparecimento dos sintomas diagnósticos iniciais (20–23).
Em até 46% dos pacientes com HELLP, mutações genéticas foram descritas nos reguladores da via alternativa do sistema complemento (24, 25). Nos estágios iniciais da gravidez, quando a placenta está se desenvolvendo, a ativação do sistema complemento é observada em níveis elevados de Bb no soro em pacientes que desenvolveram pré-eclâmpsia posteriormente (26). A ativação do sistema complemento foi observada na pré-eclâmpsia grave e na síndrome HELLP, e níveis elevados de complexo terminal (C5b-9) foram detectados na urina de pacientes com pré-eclâmpsia grave. Em pacientes com síndrome HELLP, o aumento da ativação do sistema complemento foi demonstrado por teste funcional e, além disso, por deficiências de expressão em CD55 e CD59, levando à diminuição da regulação e ativação exacerbada do sistema complemento (27-29).
Existem muitas semelhanças entre a síndrome HELLP e a SHUS. Em ambas as condições, o distúrbio do endotélio está claramente envolvido, seguido pela ativação do complemento e da coagulação. Na pré-eclâmpsia grave e na síndrome HELLP, o eculizumabe administrado nos primeiros sinais de doença grave e TMA seria benéfico para a proteção dos rins e do endotélio materno (30). Preservar a integridade endotelial pode potencialmente proteger o paciente de riscos à saúde a longo prazo, como doenças cardiovasculares. Normalmente, em TMAs relacionados à gravidez, os achados clínicos de hemólise e trombocitopenia remitem em ~ 3 dias. Se a atividade da doença durar mais, o diagnóstico diferencial deve ser continuado e somente então o tratamento alternativo (por exemplo, eculizumabe) é considerado. A introdução do tratamento com eculizumab mais cedo beneficiaria as mulheres, prevenindo danos renais e minimizando a turbulência no endotélio e a inflamação sistêmica. O eculizumabe, administrado precocemente e em apenas uma ou duas doses, pode ser suficiente para interromper a turbulência e interromper a cascata de eventos (31). Por um lado, o eculizumabe é um medicamento muito caro, mas por outro lado, o custo do tratamento intensivo, plasma, hemodiálise, possível transplante renal, sem falar nas consequências emocionais para as mães e familiares permanecem inestimáveis.
Em conclusão, nosso paciente estava gravemente doente, apresentando hemólise, distúrbios na coagulação, danos ao fígado e insuficiência renal, necessitando de internação na UTI e hemodiálise. É possível que o eculizumabe, se iniciado mais cedo, aos primeiros sinais da síndrome HELLP, possa ter sido benéfico durante o curso tardio da doença, potencialmente mitigando a lesão renal e, portanto, evitando a necessidade de hemodiálise e, posteriormente, DRC. Portanto, nesta era da medicina imunológica moderna, poderíamos fazer mais pelas mães e famílias do que apenas esperar e torcer pelo melhor?
Declaração de ética
A revisão e a aprovação éticas não foram necessário para o estudo em participantes humanos de acordo com a legislação local e requisitos institucionais. Os pacientes / participantes forneceram seu consentimento informado por escrito para participar deste estudo.
Contribuições dos autores
AL e JH-E desenvolveram a ideia para o manuscrito e escreveram o manuscrito. JH-E e MH estiveram envolvidos no cuidado diagnóstico e terapêutico do paciente. Todos os autores revisaram e editaram o manuscrito e aprovaram sua versão final para publicação.
Conflito de interesses
Os autores declaram que a pesquisa foi conduzida na ausência de quaisquer relações comerciais ou financeiras que pode ser interpretado como um potencial conflito de interesses.
4. Hauspurg A, Ying W, Hubel CA, Michos ED, Ouyang P. Resultados adversos da gravidez e futuras doenças cardiovasculares maternas. Clin Cardiol. (2018) 41: 239–46. doi: 10.1002 / clc.22887
Resumo PubMed | CrossRef Full Text | Google Scholar
5. Brosens I, Pijnenborg R, Vercruysse L, Romero R. The ” grandes Síndromes Obstétricas “estão associadas a distúrbios de placentação profunda. Am J Obstet Gynecol. (2011) 204: 193-201. doi: 10.1016 / j.ajog.2010.08.009
Resumo PubMed | CrossRef Full Text | Google Scholar
8. Germain SJ, Sacks GP, Soorana SR, Sargent IL, Redman CW . Priming inflamatório sistêmico na gravidez normal e pré-eclâmpsia: o papel das micropartículas de sinciciotrofoblasto circulantes. J Immunol. (2007) 178: 5949–56. doi: 10.4049 / jimmunol.178.9.5949
Resumo PubMed | CrossRef Full Text | Google Scholar
9. Tannetta D, Masliukaite I, Vatish M, Redman C, Sargent I Atualização de vesículas extracelulares derivadas de sinciciotrofoblasto na gravidez normal e pré-eclâmpsia. J Reprod Immunol. (2017) 119: 98–106. doi: 10.1016 / j.jri.2016.08.008
Resumo PubMed | CrossRef Full Text | Google Scholar
10. Weinstein L. Síndrome de hemólise, enzimas hepáticas elevadas e plaquetas baixas contagem: uma consequência grave da hipertensão na gravidez. Am J Obstet Gynecol. (1982) 142: 159–67. doi: 10.1016 / S0002-9378 (16) 32330-4
Resumo PubMed | CrossRef Full Text | Google Scholar
15. Pourrat O, Coudroy R, Pierre F. Diferenciação entre a síndrome HELLP grave e microangiopatia trombótica, púrpura trombocitopênica trombótica e outros imitadores. Eur J Obstet Gynecol Reprod Biol. (2015) 189: 68–72. doi: 10.1016 / j.ejogrb.2015.03.017
Resumo PubMed | CrossRef Full Text | Google Scholar
20. Agatisa PK, Ness RB, Roberts JM, Costantino JP, Kuller LH , McLaughlin MK. Comprometimento da função endotelial em mulheres com história de pré-eclâmpsia: um indicador de risco cardiovascular. Am J Physiol Hear Circ Physiol. (2004) 286: 1–3. doi: 10.1152 / ajpheart.00298.2003
Resumo PubMed | CrossRef Full Text | Google Scholar
22. Kvehaugen AS, Dechend R, Ramstad HB, Troisi R, Fugelseth D , Equipe AC. A função endotelial e os biomarcadores circulantes são alterados em mulheres e crianças após a pré-eclâmpsia. Hipertensão. (2011) 58: 63–9. doi: 10.1161 / HYPERTENSIONAHA.111.172387
Resumo PubMed | CrossRef Full Text | Google Scholar
23. Stanhewicz AE. Disfunção vascular residual em mulheres com história de pré-eclâmpsia. Am J Physiol Regul Integr Comp Physiol. (2018) 315: R1062 – R71. doi: 10.1152 / ajpregu.00204.2018
Resumo PubMed | CrossRef Full Text | Google Scholar
25. Vaught AJ, Braunstein EM, Jasem J, Yuan X, Makhlin I Eloundou S, et al. Mutações na linha germinativa na via alternativa do complemento predispõem à síndrome HELLP. Visão da JCI. (2018) 3: 0–13. doi: 10.1172 / jci.insight.99128
Resumo PubMed | CrossRef Full Text | Google Scholar
31. Elabd H, Elkholi M, Steinberg L, Acharya A. Eculizumab, um novo potencial tratamento para lesão renal aguda associada à pré-eclâmpsia / síndrome HELLP. BMJ Case Rep. (2019) 12: e228709. doi: 10.1136 / bcr-2018-228709
Resumo PubMed | CrossRef Full Text | Google Scholar