Fronteiras na genética
Introdução
Os promotores são os elementos-chave que pertencem às regiões não codificantes do genoma. Eles controlam amplamente a ativação ou repressão dos genes. Eles estão localizados perto e a montante do local de início da transcrição do gene (TSS). A região flanqueadora do promotor de um gene pode conter muitos elementos e motivos curtos de DNA (5 e 15 bases de comprimento) que servem como locais de reconhecimento para as proteínas que fornecem iniciação e regulação adequadas da transcrição do gene downstream (Juven-Gershon et al., 2008). A iniciação da transcrição gênica é a etapa mais fundamental na regulação da expressão gênica. O núcleo do promotor é um trecho mínimo de sequência de DNA que conaciona o TSS e é suficiente para iniciar diretamente a transcrição. O comprimento do promotor do núcleo varia tipicamente entre 60 e 120 pares de bases (bp).
A caixa TATÁ é uma subsequência do promotor que indica a outras moléculas onde a transcrição começa. Foi denominado “TATA-box”, pois sua sequência é caracterizada pela repetição de pares de bases T e A (TATAAA) (Baker et al., 2003). A grande maioria dos estudos sobre a TATA-box foi realizada em humanos, leveduras, e genomas de Drosophila, no entanto, elementos semelhantes foram encontrados em outras espécies, como arquéias e eucariotos antigos (Smale e Kadonaga, 2003). No caso humano, 24% dos genes têm regiões promotoras contendo TATA-box (Yang et al., 2007 ). Em eucariotos, a TATA-box está localizada a ~ 25 bp a montante do TSS (Xu et al., 2016). É capaz de definir a direção da transcrição e também indica a fita de DNA a ser lida. Proteínas chamadas de fatores de transcrição ligam-se a várias regiões não codificantes, incluindo TATA-box e recrutam uma enzima chamada RNA polimerase, que sintetiza RNA a partir do DNA.
Devido ao importante papel dos promotores na transcrição do gene, a previsão precisa dos locais promotores torna-se uma etapa necessária na expressão gênica, interpretação de padrões e construção e compreensão ção da funcionalidade das redes de regulação genética. Houve diferentes experimentos biológicos para identificação de promotores, como análise de mutação (Matsumine et al., 1998) e ensaios de imunoprecipitação (Kim et al., 2004; Dahl e Collas, 2008). No entanto, esses métodos eram caros e demorados. Recentemente, com o desenvolvimento do sequenciamento de próxima geração (NGS) (Behjati e Tarpey, 2013), mais genes de diferentes organismos foram sequenciados e seus elementos gênicos foram explorados computacionalmente (Zhang et al., 2011). Por outro lado, a inovação da tecnologia NGS resultou em uma queda dramática do custo de todo o sequenciamento do genoma, portanto, mais dados de sequenciamento estão disponíveis. A disponibilidade de dados atrai pesquisadores para desenvolver modelos computacionais para a tarefa de previsão do promotor. No entanto, ainda é uma tarefa incompleta e não há nenhum software eficiente que possa prever com precisão os promotores.
Os preditores do promotor podem ser categorizados com base na abordagem utilizada em três grupos: abordagem baseada em sinal e abordagem baseada em conteúdo e a abordagem baseada em GpG. Os preditores baseados em sinal se concentram em elementos promotores relacionados ao sítio de ligação da RNA polimerase e ignoram as porções não-elemento da sequência. Como resultado, a precisão da previsão era fraca e não satisfatória. Exemplos de preditores baseados em sinal incluem: PromoterScan (Prestridge, 1995) que usou as características extraídas da TATA-box e uma matriz ponderada de sítios de ligação de fator de transcrição com um discriminador linear para classificar as sequências promotoras das não promotoras; Promoter2.0 (Knudsen, 1999) que extraiu os recursos de diferentes caixas, como TATA-Box, CAAT-Box e GC-Box, e os passou para redes neurais artificiais (RNA) para classificação; NNPP2.1 (Reese, 2001) que utilizou o elemento iniciador (Inr) e TATA-Box para extração de características e uma rede neural de atraso de tempo para classificação, e Down e Hubbard (2002) que usou TATA-Box e utilizou máquinas de vetor de relevância (RVM) como um classificador. Os preditores baseados em conteúdo contavam com a frequência de k-mer executando uma janela de comprimento de k na sequência. No entanto, esses métodos ignoram as informações espaciais dos pares de bases nas sequências. Exemplos de preditores baseados em conteúdo incluem: PromFind (Hutchinson, 1996) que usou a frequência k-mer para realizar a previsão do promotor hexâmero; PromoterInspector (Scherf et al., 2000), que identificou as regiões contendo promotores com base em um contexto genômico comum de promotores de polimerase II por varredura para características específicas definidas como motivos de comprimento variável; MCPromoter1.1 (Ohler et al., 1999) que usou uma única cadeia de Markov interpolada (IMC) de 5ª ordem para prever sequências de promotor.Finalmente, os preditores baseados em GpG utilizaram a localização das ilhas GpG como a região promotora ou a região do primeiro exon nos genes humanos geralmente contém ilhas GpG (Ioshikhes e Zhang, 2000; Davuluri et al., 2001; Lander et al., 2001; Ponger e Mouchiroud, 2002). No entanto, apenas 60% dos promotores contêm ilhas GpG, portanto, a precisão de predição deste tipo de preditores nunca excedeu 60%.
Recentemente, abordagens baseadas em sequência foram utilizadas para predição de promotor. Yang et al. (2017) utilizou diferentes estratégias de extração de recursos para capturar as informações de sequência mais relevantes a fim de prever as interações potenciador-promotor. Lin et al. (2017) propuseram um preditor baseado em sequência, denominado “iPro70-PseZNC”, para identificação do promotor sigma70 no procarioto. Da mesma forma, Bharanikumar et al. (2018) propôs o PromoterPredict a fim de prever a força dos promotores de Escherichia coli com base em uma abordagem de regressão múltipla dinâmica em que as sequências foram representadas como matrizes de peso de posição (PWM). Kanhere e Bansal (2005) utilizaram as diferenças na estabilidade da sequência de DNA entre as sequências promotoras e não promotoras para distingui-las. Xiao et al. (2018) introduziu um preditor de duas camadas denominado iPSW (2L) -PseKNC para identificação de sequências de promotores, bem como a força dos promotores, extraindo características híbridas das sequências.
Todos os preditores mencionados acima requerem domínio- conhecimento para criar os recursos manualmente. Por outro lado, abordagens baseadas em aprendizagem profunda permitem construir modelos mais eficientes usando dados brutos (sequências de DNA / RNA) diretamente. A rede neural convolucional profunda alcançou resultados de última geração em tarefas desafiadoras, como processamento de imagem, vídeo, áudio e fala (Krizhevsky et al., 2012; LeCun et al., 2015; Schmidhuber, 2015; Szegedy et al. , 2015). Além disso, foi aplicado com sucesso em problemas biológicos como DeepBind (Alipanahi et al., 2015), DeepCpG (Angermueller et al., 2017), seleção de ponto de ramificação (Nazari et al., 2018), previsão de locais de splicing alternativos (Oubounyt et al., 2018), 2 “- predição de sítios de Ometilação (Tahir et al., 2018), quantificação de sequência de DNA (Quang e Xie, 2016), localização subcelular de proteína humana (Wei et al., 2018), etc. Além disso, A CNN recentemente ganhou atenção significativa na tarefa de reconhecimento do promotor. Muito recentemente, Umarov e Solovyev (2017) introduziram o CNNprom para a discriminação de sequências promotoras curtas. Essa arquitetura baseada na CNN alcançou resultados elevados na classificação de sequências promotoras e não promotoras. Posteriormente, este modelo foi aprimorado por Qian et al. (2018) onde os autores usaram o classificador de máquina de vetores de suporte (SVM) para inspecionar os elementos mais importantes da sequência do promotor. Esse processo resultou em melhor desempenho. Recentemente, o modelo de identificação de promotor longo foi proposto por Umarov et al. (2019) em que os autores se concentraram na identificação da posição do TSS.
Em todos os trabalhos citados, o conjunto negativo foi extraído de regiões não promotoras do genoma. Sabendo que as sequências do promotor são ricas exclusivamente em elementos funcionais específicos, como TATA-box que está localizada em –30 ~ –25 bp, GC-Box que está localizada em –110 ~ –80 bp, CAAT-Box que está localizada em – 80 ~ –70 bp, etc. Isso resulta em alta precisão de classificação devido à enorme disparidade entre as amostras positivas e negativas em termos de estrutura de sequência. Além disso, a tarefa de classificação torna-se fácil para atingir, por exemplo, os modelos da CNN contarão apenas com a presença ou ausência de alguns motivos em suas posições específicas para tomar a decisão sobre o tipo de sequência. Assim, esses modelos têm precisão / sensibilidade muito baixa (alto falso positivo) quando são testados em sequências genômicas que possuem motivos promotores, mas não são sequências promotoras. É bem sabido que existem mais motivos TATAAA no genoma do que os pertencentes às regiões promotoras. Por exemplo, sozinha a sequência de DNA do cromossomo humano 1, ftp://ftp.ensembl.org/pub/release-57/fasta/homo_sapiens/dna/, contém 151 656 motivos TATAAA. É mais do que o número máximo aproximado de genes no genoma humano total. Como ilustração desse problema, observamos que, ao testar esses modelos em sequências não promotoras que possuem TATA-box, eles classificam incorretamente a maioria dessas sequências. Portanto, para gerar um classificador robusto, o conjunto negativo deve ser selecionado com cuidado, pois determina as características que serão utilizadas pelo classificador para discriminar as classes. A importância desta ideia foi demonstrada em trabalhos anteriores, como (Wei et al., 2014). Neste trabalho, abordamos principalmente esta questão e propomos uma abordagem que integra alguns dos motivos funcionais da classe positiva na classe negativa para reduzir a dependência do modelo em relação a esses motivos.Utilizamos um CNN combinado com o modelo LSTM para analisar as características da sequência de promotores eucarióticos TATÁ e não TATÁ humanos e de camundongo e construir modelos computacionais que podem discriminar com precisão sequências promotoras curtas de não promotoras.
Materiais e métodos
2.1. Conjunto de dados
Os conjuntos de dados, que são usados para treinar e testar o preditor de promotor proposto, são coletados de humanos e camundongos. Eles contêm duas classes distintas de promotores, nomeadamente promotores TATA (isto é, as sequências que contêm TATA-box) e promotores não TATA. Esses conjuntos de dados foram construídos a partir do Eukaryotic Promoter Database (EPDnew) (Dreos et al., 2012). O EPDnew é uma nova seção sob o conhecido conjunto de dados EPD (Périer et al., 2000) que é anotado como uma coleção não redundante de promotores POL II eucarióticos onde o local de início da transcrição foi determinado experimentalmente. Ele fornece promotores de alta qualidade em comparação com a coleção de promotores ENSEMBL (Dreos et al., 2012) e está publicamente acessível em https://epd.epfl.ch//index.php. Baixamos as sequências genômicas do promotor TATA e não TATA para cada organismo de EPDnew. Esta operação resultou na obtenção de quatro conjuntos de dados de promotores, a saber: Human-TATA, Human-não-TATA, Mouse-TATA e Mouse-non-TATA. Para cada um desses conjuntos de dados, um conjunto negativo (sequências não promotoras) com o mesmo tamanho do positivo é construído com base na abordagem proposta, conforme descrito na seção a seguir. Os detalhes sobre o número de sequências promotoras para cada organismo são dados na Tabela 1. Todas as sequências têm um comprimento de 300 bp e foram extraídas de -249 ~ + 50 bp (+1 refere-se à posição TSS). Como um controle de qualidade, usamos a validação cruzada de 5 vezes para avaliar o modelo proposto. Nesse caso, 3 dobras são usadas para treinamento, 1 dobra é usado para validação e a dobra restante é usada para teste. Assim, o modelo proposto é treinado 5 vezes e o desempenho geral do 5 vezes é calculado.

Tabela 1. Estatísticas dos quatro conjuntos de dados usados neste estudo.
2.2. Construção do conjunto de dados negativo
Para treinar um modelo que possa realizar com precisão a classificação de sequências promotoras e não promotoras, precisamos escolher o conjunto negativo (sequências não promotoras) com cuidado. Este ponto é crucial para fazer um modelo capaz de generalizar bem e, portanto, capaz de manter sua precisão quando avaliado em conjuntos de dados mais desafiadores. Trabalhos anteriores, como (Qian et al., 2018), construíram um conjunto negativo selecionando aleatoriamente fragmentos de regiões não promotoras do genoma. Obviamente, essa abordagem não é totalmente razoável, porque se não houver interseção entre conjuntos positivos e negativos. Assim, o modelo encontrará facilmente recursos básicos para separar as duas classes. Por exemplo, o motivo TATA pode ser encontrado em todas as sequências positivas em uma posição específica (normalmente 28 bp a montante do TSS, entre –30 e –25 pb em nosso conjunto de dados). Portanto, criar um conjunto negativo aleatoriamente que não contenha esse motivo produzirá alto desempenho neste conjunto de dados. No entanto, o modelo falha em classificar as sequências negativas que têm o motivo TATA como promotores. Em resumo, a principal falha dessa abordagem é que ao treinar um modelo de aprendizado profundo, ele apenas aprende a discriminar as classes positivas e negativas com base na presença ou ausência de alguns recursos simples em posições específicas, o que torna esses modelos impraticáveis. Neste trabalho, pretendemos resolver este problema estabelecendo um método alternativo para derivar o conjunto negativo do positivo.
O nosso método baseia-se no facto de sempre que as características são comuns entre o negativo e o classe positiva o modelo tende, ao tomar a decisão, a ignorar ou reduzir sua dependência desses recursos (ou seja, atribuir pesos baixos a esses recursos). Em vez disso, o modelo é forçado a buscar recursos mais profundos e menos óbvios. Modelos de aprendizado profundo geralmente sofrem de convergência lenta durante o treinamento neste tipo de dados. No entanto, este método melhora a robustez do modelo e garante generalização. Reconstruímos o conjunto negativo da seguinte maneira. Cada sequência positiva gera uma sequência negativa. A sequência positiva é dividida em 20 subsequências. Então, 12 subsequências são escolhidas aleatoriamente e substituídas aleatoriamente. As 8 subsequências restantes são conservadas. Esse processo é ilustrado na Figura 1. A aplicação desse processo ao conjunto positivo resulta em novas sequências não promotoras com partes conservadas das sequências promotoras (as subsequências inalteradas, 8 subsequências de 20). Esses parâmetros permitem gerar um conjunto negativo que possui 32 e 40% de suas sequências contendo porções conservadas de sequências promotoras. Esta proporção é considerada ideal para ter um preditor de promotor robusto, conforme explicado na seção 3.2.Como as partes conservadas ocupam as mesmas posições nas sequências negativas, os motivos óbvios, como TATA-box e TSS, são agora comuns entre os dois conjuntos com uma proporção de 32 ~ 40%. Os logotipos de sequência dos conjuntos positivo e negativo para os dados do promotor TATA humano e de camundongo são mostrados nas Figuras 2, 3, respectivamente. Pode-se observar que os conjuntos positivo e negativo compartilham os mesmos motivos básicos nas mesmas posições, como o motivo TATA na posição -30 e –25 bp e o TSS na posição +1 bp. Portanto, o treinamento é mais desafiador, mas o modelo resultante generaliza bem.
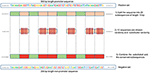
Figura 1. Ilustração do método de construção de conjunto negativo. Verde representa as subsequências conservadas aleatoriamente, enquanto vermelho representa aquelas escolhidas aleatoriamente e substituídas.

Figura 2. O logotipo da sequência no promotor TATA humano para o conjunto positivo (A) e negativo (B). Os gráficos mostram a conservação dos motivos funcionais entre os dois conjuntos.

Figura 3. O logotipo da sequência no promotor TATA do mouse para o conjunto positivo (A) e o conjunto negativo (B). Os gráficos mostram a conservação dos motivos funcionais entre os dois conjuntos.
2.3. Os modelos propostos
Propomos um modelo de aprendizado profundo que combina camadas de convolução com camadas recorrentes, conforme mostrado na Figura 4. Ele aceita uma única sequência genômica bruta, S = {N1, N2,…, Nl} onde N ∈ {A, C, G, T} e l é o comprimento da sequência de entrada, como entrada e saída de uma pontuação com valor real. A entrada é codificada em um ponto e representada como um vetor unidimensional com quatro canais. O comprimento do vetor l = 300 e os quatro canais são A, C, G e T e representados como (1 0 0 0), (0 1 0 0), (0 0 1 0), (0 0 0 1 ), respectivamente. Para selecionar o modelo de melhor desempenho, usamos o método de pesquisa de grade para escolher os melhores hiperparâmetros. Tentamos diferentes arquiteturas, como CNN sozinho, LSTM sozinho, BiLSTM sozinho, CNN combinado com LSTM. Os hiperparâmetros ajustados são o número de camadas de convolução, tamanho do kernel, número de filtros em cada camada, o tamanho da camada de pooling máxima, probabilidade de dropout e as unidades da camada Bi-LSTM.
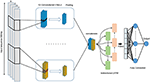
Figura 4. A arquitetura do modelo DeePromoter proposto.
O modelo proposto começa com múltiplas camadas de convolução que são alinhadas em paralelo e ajudam a aprender os motivos importantes das sequências de entrada com diferentes tamanhos de janela. Usamos três camadas de convolução para promotores não TATA com tamanhos de janela de 27, 14 e 7, e duas camadas de convolução para promotores TATA com tamanhos de janela de 27, 14. Todas as camadas de convolução são seguidas pela função de ativação ReLU (Glorot et al. , 2011), uma camada de pooling máxima com um tamanho de janela de 6 e uma camada de dropout de probabilidade 0,5. Em seguida, as saídas dessas camadas são concatenadas e alimentadas em uma camada de memória de curto prazo longa bidirecional (BiLSTM) (Schuster e Paliwal, 1997) com 32 nós para capturar as dependências entre os motivos aprendidos das camadas de convolução. Os recursos aprendidos após BiLSTM são nivelados e seguidos por abandono com uma probabilidade de 0,5. Em seguida, adicionamos duas camadas totalmente conectadas para classificação. O primeiro possui 128 nós e é seguido por ReLU e dropout com uma probabilidade de 0,5 enquanto a segunda camada é usada para predição com um nó e função de ativação sigmóide. BiLSTM permite que a informação persista e aprenda dependências de longo prazo de amostras sequenciais como DNA e RNA. Isso é obtido por meio da estrutura LSTM, que é composta por uma célula de memória e três portas chamadas portas de entrada, saída e esquecer. Essas portas são responsáveis por regular as informações na célula de memória. Além disso, a utilização do módulo LSTM aumenta a profundidade da rede, enquanto o número de parâmetros necessários permanece baixo. Ter uma rede mais profunda permite extrair características mais complexas e este é o objetivo principal de nossos modelos, pois o conjunto negativo contém amostras duras.
O framework Keras é usado para construir e treinar os modelos propostos (Chollet F. et al., 2015). O otimizador Adam (Kingma e Ba, 2014) é usado para atualizar os parâmetros com uma taxa de aprendizado de 0,001. O tamanho do lote é definido como 32 e o número de épocas é definido como 50. A parada antecipada é aplicada com base na perda de validação.
Resultados e discussão
3.1. Medidas de desempenho
Neste trabalho, usamos as métricas de avaliação amplamente adotadas para avaliar o desempenho dos modelos propostos.Essas métricas são precisão, recall e coeficiente de correlação de Matthew (MCC), e são definidas da seguinte forma:
Onde TP é verdadeiro positivo e representa sequências promotoras corretamente identificadas, TN é verdadeiro negativo e representa sequências promotoras rejeitadas corretamente, FP é falso positivo e representa sequências identificadas incorretamente sequências promotoras, e FN é falso negativo e representa sequências promotoras rejeitadas incorretamente.
3.2. Efeito do Conjunto Negativo
Ao analisar os trabalhos publicados anteriormente para identificação de sequências promotoras, notamos que o desempenho desses trabalhos depende muito da forma de preparação do conjunto de dados negativos. Eles tiveram um desempenho muito bom nos conjuntos de dados que prepararam, no entanto, eles têm uma alta proporção de falsos positivos quando avaliados em um conjunto de dados mais desafiador que inclui sequências não prompter com motivos comuns com sequências promotoras. Por exemplo, no caso do conjunto de dados do promotor TATA, as sequências geradas aleatoriamente não terão motivo TATA na posição -30 e –25 bp, o que, por sua vez, torna a tarefa de classificação mais fácil. Em outras palavras, seu classificador dependia da presença do motivo TATA para identificar a sequência do promotor e, como resultado, era fácil obter um alto desempenho nos conjuntos de dados que prepararam. No entanto, seus modelos falharam drasticamente ao lidar com sequências negativas que continham o motivo TATA (exemplos difíceis). A precisão diminuiu conforme a taxa de falsos positivos aumentava. Simplesmente, eles classificaram essas sequências como sequências promotoras positivas. Uma análise semelhante é válida para os outros motivos promotores. Portanto, o objetivo principal de nosso trabalho não é apenas alcançar alto desempenho em um conjunto de dados específico, mas também aprimorar a capacidade do modelo em generalizar bem, treinando em um conjunto de dados desafiador.
Para ilustrar melhor este ponto, treinamos e testar nosso modelo nos conjuntos de dados do promotor TATA humano e de camundongo com diferentes métodos de preparação de conjuntos negativos. O primeiro experimento é realizado usando sequências negativas amostradas aleatoriamente de regiões não codificantes do genoma (ou seja, semelhante à abordagem usada nos trabalhos anteriores). Notavelmente, nosso modelo proposto atinge uma precisão de predição quase perfeita (precisão = 99%, recall = 99%, Mcc = 98%) e (precisão = 99%, recall = 98%, Mcc = 97%) para humanos e camundongos, respectivamente . Esses resultados elevados são esperados, mas a questão é se esse modelo pode manter o mesmo desempenho quando avaliado em um conjunto de dados que tem exemplos difíceis. A resposta, com base na análise dos modelos anteriores, é não. O segundo experimento é realizado usando nosso método proposto para preparar o conjunto de dados conforme explicado na seção 2.2. Preparamos os conjuntos negativos que contêm TATA-box conservado com diferentes percentagens, como 12, 20, 32 e 40% e o objetivo é reduzir a lacuna entre a precisão e o recall. Isso garante que nosso modelo aprenda recursos mais complexos em vez de aprender apenas a presença ou ausência de TATA-box. Conforme mostrado nas Figuras 5A, B, o modelo estabiliza na proporção de 32 ~ 40% para conjuntos de dados do promotor TATA humano e de camundongo.
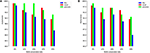
Figura 5. O efeito de diferentes razões de conservação do motivo TATÁ no conjunto negativo sobre o desempenho no caso do conjunto de dados do promotor TATÁ para humanos (A) e camundongos (B) .
3.3. Resultados e comparação
Nos últimos anos, muitas ferramentas de previsão da região promotora foram propostas (Hutchinson, 1996; Scherf et al., 2000; Reese, 2001; Umarov e Solovyev, 2017). No entanto, algumas dessas ferramentas não estão publicamente disponíveis para teste e algumas delas requerem mais informações além das sequências genômicas brutas. Neste estudo, comparamos o desempenho de nossos modelos propostos com o trabalho atual do estado da arte, CNNProm, que foi proposto por Umarov e Solovyev (2017) conforme mostrado na Tabela 2. Em geral, os modelos propostos, DeePromoter, claramente superam o CNNProm em todos os conjuntos de dados com todas as métricas de avaliação. Mais especificamente, o DeePromoter melhora a precisão, a recuperação e o MCC no caso do conjunto de dados TATA humano em 0,18, 0,04 e 0,26, respectivamente. No caso do conjunto de dados não-TATÁ humano, o DeePromoter melhora a precisão em 0,39, o recall em 0,12 e o MCC em 0,66. Da mesma forma, o DeePromoter melhora a precisão e o MCC no caso do conjunto de dados TATA do mouse em 0,24 e 0,31, respectivamente. No caso do conjunto de dados não-TATA do mouse, o DeePromoter melhora a precisão em 0,37, o recall em 0,04 e o MCC em 0,65. Esses resultados confirmam que o CNNProm falha em rejeitar sequências negativas com o promotor TATA, portanto, tem altos falsos positivos. Por outro lado, nossos modelos são capazes de lidar com esses casos com mais sucesso e a taxa de falsos positivos é menor em comparação com o CNNProm.

Tabela 2. Comparação do DeePromoter com o estado de método da arte.
Para análises adicionais, estudamos o efeito da alternância de nucleotídeos em cada posição na pontuação de saída. Focamos na região –40 e 10 bp, pois hospeda a parte mais importante da sequência do promotor. Para cada sequência de promotor no conjunto de teste, realizamos varredura de mutação computacional para avaliar o efeito da mutação de cada base da subsequência de entrada (150 substituições no intervalo –40 ~ 10 bp subsequência). Isso é ilustrado nas Figuras 6, 7 para conjuntos de dados TATA humanos e de camundongo, respectivamente. A cor azul representa uma queda na pontuação de saída devido à mutação, enquanto a cor vermelha representa o incremento da pontuação devido à mutação. Notamos que alterar os nucleotídeos para C ou G na região –30 e –25 bp reduz a pontuação de saída significativamente. Esta região é TATA-box que é um motivo funcional muito importante na sequência do promotor. Assim, nosso modelo é capaz de encontrar com sucesso a importância desta região. No resto das posições, os nucleotídeos C e G são mais preferíveis do que A e T, especialmente no caso do camundongo. Isso pode ser explicado pelo fato de que a região promotora tem mais nucleotídeos C e G do que A e T (Shi e Zhou, 2006).

Figura 6. O mapa de saliência da região –40 bp a 10 bp, que inclui a TATA-box, no caso de sequências do promotor TATA humano.

Figura 7. O mapa de saliência da região –40 bp a 10 bp, que inclui a caixa TATA, no caso de sequências do promotor TATA de camundongo.
Conclusão
A predição precisa das sequências do promotor é essencial para a compreensão do mecanismo subjacente do processo de regulação do gene. Neste trabalho, desenvolvemos DeePromoter – que é baseado em uma combinação de rede neural de convolução e LSTM bidirecional – para prever as sequências de promotor de eucariotos curtos no caso de humanos e camundongos para os promotores TATA e não TATA. O componente essencial deste trabalho foi superar o problema da baixa precisão (alta taxa de falsos positivos) observada nas ferramentas desenvolvidas anteriormente devido à dependência de algumas características / motivos óbvios na sequência ao classificar sequências promotoras e não promotoras. Neste trabalho, estávamos particularmente interessados em construir um conjunto negativo rígido que leva os modelos a explorar a sequência para características profundas e relevantes, em vez de apenas distinguir as sequências promotoras e não promotoras com base na existência de alguns motivos funcionais. Os principais benefícios de usar o DeePromoter é que ele reduz significativamente o número de previsões de falsos positivos, ao mesmo tempo que atinge alta precisão em conjuntos de dados desafiadores. O DeePromoter superou o método anterior não apenas no desempenho, mas também na superação do problema de altas previsões de falsos positivos. Projeta-se que esta estrutura pode ser útil em aplicações relacionadas a drogas e na academia.
Contribuições dos autores
MO e ZL prepararam o conjunto de dados, conceberam o algoritmo e realizaram o experimento e análise. MO e HT prepararam o servidor web e escreveram o manuscrito com o apoio de ZL e KC. Todos os autores discutiram os resultados e contribuíram para o manuscrito final.
Financiamento
Esta pesquisa foi apoiada pelo Programa de Pesquisa do Cérebro da Fundação Nacional de Pesquisa (NRF) financiado pelo governo coreano ( MSIT) (No. NRF-2017M3C7A1044815).
Declaração de conflito de interesses
Os autores declaram que a pesquisa foi conduzida na ausência de quaisquer relações comerciais ou financeiras que possam ser interpretadas como um potencial conflito de interesses.
Bharanikumar, R., Premkumar, KAR, and Palaniappan, A. (2018). Promoterpredict: a modelagem baseada em sequência da força do promotor σ70 de escherichia coli produz uma dependência logarítmica entre a força do promotor e a sequência. PeerJ 6: e5862. doi: 10.7717 / peerj.5862
Resumo PubMed | CrossRef Full Text | Google Scholar
Glorot, X., Bordes, A., e Bengio, Y. (2011). “Deep sparse retifier neural networks”, em Proceedings of the Fourteenth International Conference on Artificial Intelligence and Statistics, (Fort Lauderdale, FL:) 315-323.
Google Scholar
Hutchinson, G. (1996). A predição de regiões promotoras de vertebrados usando análise de frequência diferencial de hexâmeros. Bioinformática 12, 391-398.
Resumo do PubMed | Google Scholar
Kingma, DP e Ba, J. (2014). Adam: um método para otimização estocástica. arXiv preprint arXiv: 1412,6980.
Google Scholar
Knudsen, S. (1999). Promotor 2. 0: para o reconhecimento de sequências do promotor polii. Bioinformatics 15, 356-361.
Resumo do PubMed | Google Scholar
Ponger, L. e Mouchiroud, D. (2002). Cpgprod: identificação de ilhas cpg associadas a locais de início da transcrição em grandes sequências genômicas de mamíferos. Bioinformatics 18, 631–633. doi: 10.1093 / bioinformatics / 18.4.631
Resumo PubMed | CrossRef Full Text | Google Scholar
Quang, D. e Xie, X. (2016). Danq: uma rede neural profunda híbrida convolucional e recorrente para quantificar a função de sequências de DNA. Nucleic Acids Res. 44, e107 – e107. doi: 10.1093 / nar / gkw226
Resumo do PubMed | CrossRef Full Text | Google Scholar
Umarov, R. K. e Solovyev, V. V. (2017). Reconhecimento de promotores procarióticos e eucarióticos usando redes neurais convolucionais de aprendizagem profunda. PLoS ONE 12: e0171410. doi: 10.1371 / journal.pone.0171410
PubMed Resumo | CrossRef Full Text | Google Scholar