Frontiere în genetică
Introducere
Promotorii sunt elementele cheie care aparțin regiunilor necodificate din genom. Ei controlează în mare măsură activarea sau reprimarea genelor. Acestea sunt situate în apropierea și în amonte situl de început al transcripției genei (TSS). Regiunea de flancare a promotorului unei gene poate conține multe elemente și motive ADN scurte cruciale (5 și 15 baze lungi) care servesc drept locuri de recunoaștere a proteinelor inițierea și reglarea corespunzătoare a transcripției genei din aval (Juven-Gershon și colab., 2008). Inițierea transcrierii genelor este cel mai fundamental pas în reglarea expresiei genice. Miezul promotor este o întindere minimă a secvenței ADN care conează TSS și este suficientă pentru a iniția direct transcrierea. Lungimea promotorului de bază variază de obicei între 60 și 120 de perechi de baze (bp).
Cutia TATA este o subsecvență a promotorului care indică altor molecule de unde începe transcripția. A fost denumită „cutie TATA” deoarece secvența sa se caracterizează prin repetarea perechilor de baze T și A (TATAAA) (Baker și colab., 2003). Marea majoritate a studiilor asupra cutiei TATA au fost efectuate pe drojdie umană, și genomul Drosophila, cu toate acestea, elemente similare au fost găsite la alte specii, cum ar fi archaea și eucariotele antice (Smale și Kadonaga, 2003). În cazul omului, 24% din gene au regiuni promotore care conțin TATA-box (Yang și colab., 2007 În eucariote, cutia TATA este situată la ~ 25 pb în amonte de TSS (Xu și colab., 2016). Este capabilă să definească direcția transcripției și indică, de asemenea, catena ADN care trebuie citită. Proteinele numite factori de transcripție se leagă de mai multe regiuni necodificatoare, inclusiv TATA-box și recrutează o enzimă numită ARN polimerază, care sintetizează ARN din ADN. un pas necesar în exprimarea genelor, interpretarea tiparelor și construirea și înțelegerea funcționalitatea rețelelor de reglare genetică. Au existat diferite experimente biologice pentru identificarea promotorilor, cum ar fi analiza mutațională (Matsumine și colab., 1998) și teste de imunoprecipitare (Kim și colab., 2004; Dahl și Collas, 2008). Cu toate acestea, aceste metode au fost atât costisitoare, cât și consumatoare de timp. Recent, odată cu dezvoltarea secvențierii de generație următoare (NGS) (Behjati și Tarpey, 2013) au fost secvențiate mai multe gene ale diferitelor organisme și elementele genetice ale acestora au fost explorate computerizat (Zhang și colab., 2011). Pe de altă parte, inovația tehnologiei NGS a dus la o scădere dramatică a costului secvențierii întregului genom, astfel, sunt disponibile mai multe date de secvențiere. Disponibilitatea datelor îi atrage pe cercetători să dezvolte modele de calcul pentru sarcina de predicție a promotorilor. Cu toate acestea, este încă o sarcină incompletă și nu există un software eficient care să poată prezice cu precizie promotorii.
Predictorii promotorilor pot fi clasificați pe baza abordării utilizate în trei grupuri și anume abordarea bazată pe semnal, abordarea bazată pe conținut. , și abordarea bazată pe GpG. Predictorii bazați pe semnal se concentrează pe elementele promotor legate de situsul de legare a ARN polimerazei și ignoră porțiunile neelementale ale secvenței. Ca urmare, precizia predicției a fost slabă și nu a fost satisfăcătoare. Exemple de predictori pe bază de semnal includ: PromoterScan (Prestridge, 1995) care a folosit caracteristicile extrase ale casetei TATA și o matrice ponderată a siturilor de legare a factorului de transcripție cu un discriminator liniar pentru a clasifica secvențele promotor din cele care nu sunt promotor; Promoter2.0 (Knudsen, 1999) care a extras caracteristicile din diferite cutii precum TATA-Box, CAAT-Box și GC-Box și le-a transmis rețelelor neuronale artificiale (ANN) pentru clasificare; NNPP2.1 (Reese, 2001) care a folosit elementul inițiator (Inr) și TATA-Box pentru extragerea caracteristicilor și o rețea neuronală cu întârziere pentru clasificare, și Down și Hubbard (2002) care au folosit TATA-Box și au folosit o mașină vectorială de relevanță (RVM) ca clasificator. Predictorii bazați pe conținut s-au bazat pe numărarea frecvenței k-mer prin rularea unei ferestre de lungime k în secvență. Cu toate acestea, aceste metode ignoră informațiile spațiale ale perechilor de baze din secvențe. Exemple de predictori bazate pe conținut includ: PromFind (Hutchinson, 1996) care a folosit frecvența k-mer pentru a efectua predicția promotorului hexamerului; PromoterInspector (Scherf și colab., 2000) care a identificat regiunile care conțin promotori pe baza unui context genomic comun al promotorilor polimerazei II prin scanarea caracteristicilor specifice definite ca motive cu lungime variabilă; MCPromoter1.1 (Ohler și colab., 1999) care a folosit un singur lanț Markov interpolat (IMC) de ordinul 5 pentru a prezice secvențele promotorului.În cele din urmă, predictorii pe bază de GpG au folosit locația insulelor GpG ca regiune promotor sau prima regiune exonică din genele umane conține de obicei insule GpG (Ioshikhes și Zhang, 2000; Davuluri și colab., 2001; Lander și colab., 2001; Ponger și Mouchiroud, 2002). Cu toate acestea, doar 60% dintre promotori conțin insule GpG, prin urmare precizia de predicție a acestui tip de predictori nu a depășit niciodată 60%.
Recent, abordările bazate pe secvențe au fost utilizate pentru predicția promotorului. Yang și colab. (2017) au folosit diferite strategii de extragere a caracteristicilor pentru a capta cele mai relevante informații de secvență pentru a prezice interacțiunile amplificator-promotor. Lin și colab. (2017) au propus un predictor bazat pe secvențe, numit „iPro70-PseZNC”, pentru identificarea promotorului sigma70 în procariot. La fel, Bharanikumar și colab. (2018) a propus PromoterPredict pentru a prezice puterea promotorilor Escherichia coli pe baza unei abordări dinamice de regresie multiplă în care secvențele au fost reprezentate ca matrici de greutate de poziție (PWM). Kanhere și Bansal (2005) au utilizat diferențele în stabilitatea secvenței ADN între secvențele promotor și non-promotor pentru a le distinge. Xiao și colab. (2018) a introdus un predictor cu două straturi numit iPSW (2L) -PseKNC pentru identificarea secvențelor promotorilor, precum și puterea promotorilor prin extragerea caracteristicilor hibride din secvențe.
Toți predictorii menționați anterior necesită domeniu- cunoștințe pentru a crea manual caracteristicile. Pe de altă parte, abordările bazate pe învățarea profundă permit construirea de modele mai eficiente folosind direct date brute (secvențe ADN / ARN). Rețeaua neuronală convoluțională profundă a obținut rezultate de ultimă generație în sarcini provocatoare, cum ar fi procesarea imaginii, a videoclipurilor, a sunetului și a vorbirii (Krizhevsky și colab., 2012; LeCun și colab., 2015; Schmidhuber, 2015; Szegedy și colab. , 2015). În plus, a fost aplicat cu succes în probleme biologice precum DeepBind (Alipanahi și colab., 2015), DeepCpG (Angermueller și colab., 2017), selecția punctelor ramificate (Nazari și colab., 2018), predicția alternativă a locurilor de îmbinare (Oubounyt și colab., 2018), predicția siturilor de 2 „-Ometilare (Tahir și colab., 2018), cuantificarea secvenței ADN (Quang și Xie, 2016), localizarea subcelulară a proteinelor umane (Wei și colab., 2018) etc. CNN a câștigat recent o atenție semnificativă în sarcina de recunoaștere a promotorilor. Foarte recent, Umarov și Solovyev (2017) au introdus CNNprom pentru discriminarea secvențelor de promotori scurți, această arhitectură bazată pe CNN a obținut rezultate ridicate în clasificarea secvențelor de promotori și non-promotori. Ulterior, acest model a fost îmbunătățit de Qian și colab. (2018) unde autorii au folosit clasificatorul suport vector machine (SVM) pentru a inspecta cele mai importante elemente ale secvenței promotorului. Apoi, cele mai influente elemente au fost ținute necomprimate în timp ce le comprimau pe cele mai puțin importante. Acest proces a dus la o performanță mai bună. Recent, modelul lung de identificare a promotorului a fost propus de Umarov și colab. (2019) în care autorii s-au concentrat pe identificarea poziției TSS.
În toate lucrările menționate mai sus, setul negativ a fost extras din regiunile non-promotor ale genomului. Știind că secvențele promotorului sunt bogate exclusiv în elemente funcționale specifice, cum ar fi TATA-box care este situat la –30 ~ –25 bp, GC-Box care este situat la –110 ~ –80 bp, CAAT-Box care este situat la – 80 ~ –70 bp, etc. Acest lucru are ca rezultat o precizie ridicată a clasificării datorită disparității uriașe dintre eșantioanele pozitive și negative în ceea ce privește structura secvenței. În plus, sarcina de clasificare devine ușor de realizat, de exemplu, modelele CNN se vor baza doar pe prezența sau absența unor motive la pozițiile lor specifice pentru a lua decizia cu privire la tipul secvenței. Astfel, aceste modele au o precizie / sensibilitate foarte scăzută (fals pozitiv ridicat) atunci când sunt testate pe secvențe genomice care au motive promotor, dar nu sunt secvențe promotor. Este bine cunoscut faptul că există mai multe motive TATAAA în genom decât cele aparținând regiunilor promotor. De exemplu, numai secvența ADN a cromozomului 1 uman, ftp://ftp.ensembl.org/pub/release-57/fasta/homo_sapiens/dna/, conține 151 656 motive TATAAA. Este mai mult decât numărul maxim aproximativ de gene din genomul uman total. Ca o ilustrare a acestei probleme, observăm că atunci când testăm aceste modele pe secvențe non-promotor care au cutie TATA, acestea clasifică greșit majoritatea acestor secvențe. Prin urmare, pentru a genera un clasificator robust, setul negativ trebuie selectat cu atenție, deoarece determină caracteristicile care vor fi utilizate de clasificator pentru a discrimina clasele. Importanța acestei idei a fost demonstrată în lucrări anterioare precum (Wei și colab., 2014). În această lucrare, abordăm în principal această problemă și propunem o abordare care integrează unele dintre motivele funcționale ale clasei pozitive din clasa negativă pentru a reduce dependența modelului de aceste motive.Utilizăm un CNN combinat cu modelul LSTM pentru a analiza caracteristicile secvenței promotorilor eucariotici TATA și non-TATA pentru om și șoarece și construim modele de calcul care pot discrimina cu precizie secvențele promotorului scurt de cele non-promotor.
Materiale și metode
2.1. Set de date
Seturile de date, care sunt utilizate pentru instruirea și testarea predictorului promotor propus, sunt colectate de la om și șoarece. Acestea conțin două clase distincte de promotori și anume promotori TATA (adică secvențele care conțin caseta TATA) și promotori non-TATA. Aceste seturi de date au fost construite din baza de date Eukaryotic Promoter (EPDnew) (Dreos și colab., 2012). EPDnew este o nouă secțiune din cunoscutul set de date EPD (Périer și colab., 2000), care este adnotată o colecție non-redundantă de promotori eucariote POL II în care site-ul de început al transcrierii a fost determinat experimental. Oferă promotori de înaltă calitate comparativ cu colecția de promotori ENSEMBL (Dreos și colab., 2012) și este accesibil publicului la https://epd.epfl.ch//index.php. Am descărcat secvențe genomice promotor TATA și non-TATA pentru fiecare organism din EPDnew. Această operațiune a dus la obținerea a patru seturi de date promotor și anume: Human-TATA, Human-non-TATA, Mouse-TATA și Mouse-non-TATA. Pentru fiecare dintre aceste seturi de date, un set negativ (secvențe non-promotor) cu aceeași dimensiune a celui pozitiv este construit pe baza abordării propuse așa cum este descris în secțiunea următoare. Detaliile privind numărul secvențelor promotor pentru fiecare organism sunt date în Tabelul 1. Toate secvențele au o lungime de 300 bp și au fost extrase din -249 ~ + 50 bp (+1 se referă la poziția TSS). Ca control al calității, am folosit o validare încrucișată de 5 ori pentru a evalua modelul propus. În acest caz, 3 ori sunt utilizate pentru antrenament, 1 ori este utilizat pentru validare, iar restul este utilizat pentru testare. Astfel, modelul propus este instruit de 5 ori și se calculează performanța generală de 5 ori.

Tabelul 1. Statistica celor patru seturi de date utilizate în acest studiu.
2.2. Construcția setului de date negative
Pentru a instrui un model care poate efectua cu precizie clasificarea secvențelor promotor și non-promotor, trebuie să alegem cu atenție setul negativ (secvențe non-promotor). Acest punct este crucial pentru realizarea unui model capabil să se generalizeze bine și, prin urmare, capabil să își mențină precizia atunci când este evaluat pe seturi de date mai provocatoare. Lucrările anterioare, cum ar fi (Qian și colab., 2018), au construit un set negativ selectând aleatoriu fragmente din regiunile non-promotor ale genomului. Evident, această abordare nu este complet rezonabilă, deoarece dacă nu există o intersecție între mulțimi pozitive și negative. Astfel, modelul va găsi cu ușurință caracteristici de bază pentru a separa cele două clase. De exemplu, motivul TATA poate fi găsit în toate secvențele pozitive la o poziție specifică (în mod normal 28 bp în amonte de TSS, între –30 și –25 pb în setul nostru de date). Prin urmare, crearea aleatorie a unui set negativ care nu conține acest motiv va produce performanțe ridicate în acest set de date. Cu toate acestea, modelul eșuează la clasificarea secvențelor negative care au motiv TATA drept promotori. Pe scurt, defectul major al acestei abordări este acela că, atunci când antrenează un model de învățare profundă, acesta învață doar să discrimineze clasele pozitive și negative pe baza prezenței sau absenței unor trăsături simple la poziții specifice, ceea ce face ca aceste modele să fie impracticabile. În această lucrare, ne propunem să rezolvăm această problemă prin stabilirea unei metode alternative pentru a deriva setul negativ din cel pozitiv.
Metoda noastră se bazează pe faptul că, de fiecare dată când caracteristicile sunt comune între negativ și clasă pozitivă, modelul tinde, atunci când ia decizia, să ignore sau să reducă dependența acestuia de aceste caracteristici (adică, atribuie greutăți mici acestor caracteristici). În schimb, modelul este forțat să caute caracteristici mai profunde și mai puțin evidente. Modelele de învățare profundă suferă în general de convergență lentă în timp ce se antrenează cu acest tip de date. Cu toate acestea, această metodă îmbunătățește robustețea modelului și asigură generalizarea. Reconstituim setul negativ după cum urmează. Fiecare secvență pozitivă generează o secvență negativă. Secvența pozitivă este împărțită în 20 de subsecvențe. Apoi, 12 subsecvențe sunt alese aleatoriu și substituite aleatoriu. Restul de 8 subsecvențe sunt conservate. Acest proces este ilustrat în Figura 1. Aplicarea acestui proces la setul pozitiv rezultă în noi secvențe non-promotor cu părți conservate din secvențe promotor (subsecvențele nemodificate, 8 subsecvențe din 20). Acești parametri permit generarea unui set negativ care are 32 și 40% din secvențele sale conținând porțiuni conservate de secvențe promotor. Acest raport este considerat optim pentru a avea un predictor robust al promotorului, așa cum se explică în secțiunea 3.2.Deoarece părțile conservate ocupă aceleași poziții în secvențele negative, motivele evidente, cum ar fi TATA-box și TSS, sunt acum comune între cele două seturi cu un raport de 32 ~ 40%. Logo-urile secvenței seturilor pozitive și negative atât pentru datele promotorului TATA uman, cât și pentru șoarece sunt prezentate în figurile 2, respectiv 3. Se poate vedea că seturile pozitive și negative împart aceleași motive de bază la aceleași poziții, cum ar fi motivul TATA la poziția -30 și –25 pb și TSS la poziția +1 pb. Prin urmare, instruirea este mai provocatoare, dar modelul rezultat se generalizează bine.
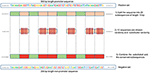
Figura 1. Ilustrarea metodei de construcție a setului negativ. Verde reprezintă subsecvențele conservate aleatoriu, în timp ce roșu reprezintă cele alese și substituite aleatoriu.

Figura 2. Logo-ul secvenței în promotorul TATA uman atât pentru setul pozitiv (A), cât și pentru setul negativ (B). Graficele arată conservarea motivelor funcționale dintre cele două seturi.

Figura 3. Logo-ul secvenței în promotorul TATA al mouse-ului atât pentru setul pozitiv (A), cât și pentru setul negativ (B). Graficele arată conservarea motivelor funcționale dintre cele două seturi.
2.3. Modelele propuse
Propunem un model de învățare profundă care combină straturile de convoluție cu straturile recurente așa cum se arată în Figura 4. Acceptă o singură secvență genomică brută, S = {N1, N2, …, Nl} unde N ∈ {A, C, G, T} și l este lungimea secvenței de intrare, ca intrare și ieșire un scor real. Intrarea este codată cu o singură temperatură și este reprezentată ca un vector unidimensional cu patru canale. Lungimea vectorului l = 300 și cele patru canale sunt A, C, G și T și sunt reprezentate ca (1 0 0 0), (0 1 0 0), (0 0 1 0), (0 0 0 1 ), respectiv. Pentru a selecta modelul cel mai performant, am folosit metoda de căutare a grilei pentru alegerea celor mai buni hiper-parametri. Am încercat diferite arhitecturi precum CNN singur, LSTM singur, BiLSTM singur, CNN combinat cu LSTM. Hiperparametrii reglați sunt numărul de straturi de convoluție, dimensiunea nucleului, numărul de filtre din fiecare strat, dimensiunea stratului maxim de grupare, probabilitatea de renunțare și unitățile stratului Bi-LSTM.
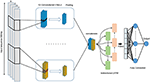
Figura 4. Arhitectura modelului DeePromoter propus.
Modelul propus începe cu mai multe straturi de convoluție care sunt aliniate în paralel și ajută la învățarea motivelor importante ale secvențelor de intrare cu dimensiuni diferite ale ferestrei. Folosim trei straturi de convoluție pentru promotorul non-TATA cu dimensiuni de ferestre de 27, 14 și 7 și două straturi de convoluție pentru promotori TATA cu dimensiuni de ferestre de 27, 14. Toate straturile de convoluție sunt urmate de funcția de activare ReLU (Glorot și colab. , 2011), un strat de pooling maxim cu o dimensiune a ferestrei de 6 și un strat dropout cu o probabilitate de 0,5. Apoi, ieșirile acestor straturi sunt concatenate împreună și introduse într-un strat de memorie bidirecțională pe termen scurt (BiLSTM) (Schuster și Paliwal, 1997) cu 32 de noduri pentru a capta dependențele dintre motivele învățate din straturile de convoluție. Caracteristicile învățate după BiLSTM sunt aplatizate și urmate de abandon cu o probabilitate de 0,5. Apoi adăugăm două straturi complet conectate pentru clasificare. Primul are 128 de noduri și este urmat de ReLU și abandon cu o probabilitate de 0,5 în timp ce al doilea strat este utilizat pentru predicție cu un nod și funcția de activare sigmoidă. BiLSTM permite informațiilor să persiste și să învețe dependențe pe termen lung de probe secvențiale, cum ar fi ADN și ARN. Acest lucru se realizează prin structura LSTM, care este compusă dintr-o celulă de memorie și trei porți numite porți de intrare, ieșire și uitare. Aceste porți sunt responsabile pentru reglarea informațiilor din celula de memorie. În plus, utilizarea modulului LSTM crește adâncimea rețelei, în timp ce numărul parametrilor necesari rămâne redus. Având o rețea mai profundă, puteți extrage caracteristici mai complexe și acesta este obiectivul principal al modelelor noastre, deoarece setul negativ conține probe dificile.
Cadrul Keras este utilizat pentru construirea și instruirea modelelor propuse (Chollet F. et. al., 2015). Optimizatorul Adam (Kingma și Ba, 2014) este utilizat pentru actualizarea parametrilor cu o rată de învățare de 0,001. Dimensiunea lotului este setată la 32 și numărul de epoci este setat la 50. Oprirea timpurie se aplică pe baza pierderii validării.
Rezultate și discuții
3.1. Măsuri de performanță
În această lucrare, folosim metricele de evaluare adoptate pe scară largă pentru evaluarea performanței modelelor propuse.Aceste valori sunt precizia, rechemarea și coeficientul de corelație Matthew (MCC) și sunt definite după cum urmează:
În cazul în care TP este adevărat pozitiv și reprezintă secvențe de promotor identificate corect, TN este adevărat negativ și reprezintă secvențe de promotor respinse corect, FP este fals pozitiv și reprezintă identificat incorect secvențe de promotor, iar FN este fals negativ și reprezintă secvențe de promotor respinse incorect.
3.2. Efectul setului negativ
Când am analizat lucrările publicate anterior pentru identificarea secvențelor promotorului, am observat că performanța acestor lucrări depinde în mare măsură de modul de pregătire a setului de date negativ. Au performat foarte bine la seturile de date pe care le-au pregătit, cu toate acestea, au un raport fals pozitiv ridicat atunci când au fost evaluați pe un set de date mai provocator, care include secvențe non-prompter având motive comune cu secvențe promotor. De exemplu, în cazul setului de date promotor TATA, secvențele generate aleatoriu nu vor avea motiv TATA la poziția -30 și –25 bp ceea ce la rândul său face sarcina clasificării mai ușoară. Cu alte cuvinte, clasificatorul lor depindea de prezența motivului TATA pentru a identifica secvența promotorului și, ca rezultat, a fost ușor să se obțină performanțe ridicate pe seturile de date pe care le-au pregătit. Cu toate acestea, modelele lor au eșuat dramatic când au avut de-a face cu secvențe negative care conțineau motiv TATA (exemple grele). Precizia a scăzut odată cu creșterea ratei fals pozitive. Pur și simplu, au clasificat aceste secvențe ca secvențe de promotor pozitive. O analiză similară este valabilă pentru celelalte motive ale promotorului. Prin urmare, scopul principal al muncii noastre nu este doar obținerea de performanțe ridicate pe un set de date specific, ci și îmbunătățirea capacității modelului de a generaliza bine prin instruirea asupra unui set de date provocator.
Pentru a ilustra mai mult acest punct, ne antrenăm și testați modelul nostru pe seturile de date promotor TATA pentru om și șoarece cu diferite metode de pregătire a seturilor negative. Primul experiment este realizat folosind secvențe negative eșantionate aleatoriu din regiuni necodificatoare ale genomului (adică, similar cu abordarea utilizată în lucrările anterioare). În mod remarcabil, modelul nostru propus atinge o precizie de predicție aproape perfectă (precizie = 99%, reamintire = 99%, Mcc = 98%) și (precizie = 99%, reamintire = 98%, Mcc = 97%) atât pentru om, cât și pentru mouse, respectiv . Se așteaptă aceste rezultate ridicate, dar întrebarea este dacă acest model poate menține aceeași performanță atunci când este evaluat pe un set de date care are exemple dificile. Răspunsul, bazat pe analiza modelelor anterioare, este nu. Al doilea experiment este realizat folosind metoda propusă pentru pregătirea setului de date, așa cum se explică în secțiunea 2.2. Pregătim seturile negative care conțin caseta TATA conservată cu procente diferite, cum ar fi 12, 20, 32 și 40%, iar obiectivul este reducerea decalajului dintre precizie și rechemare. Acest lucru asigură că modelul nostru învață caracteristici mai complexe decât să învețe doar prezența sau absența TATA-box. Așa cum se arată în figurile 5A, modelul B se stabilizează la un raport de 32 ~ 40% atât pentru seturile de date promotor TATA umane, cât și pentru șoareci.
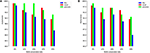
Figura 5. Efectul diferitelor rapoarte de conservare a motivului TATA în setul negativ asupra performanței în cazul setului de date promotor TATA atât pentru om (A), cât și pentru mouse (B) .
3.3. Rezultate și comparație
În ultimii ani, au fost propuse o mulțime de instrumente de predicție a regiunii promotorilor (Hutchinson, 1996; Scherf și colab., 2000; Reese, 2001; Umarov și Solovyev, 2017). Cu toate acestea, unele dintre aceste instrumente nu sunt disponibile public pentru testare și unele dintre ele necesită mai multe informații în afară de secvențele genomice brute. În acest studiu, comparăm performanța modelelor noastre propuse cu lucrarea de ultimă generație actuală, CNNProm, care a fost propusă de Umarov și Solovyev (2017) așa cum se arată în Tabelul 2. În general, modelele propuse, DeePromoter, depășește în mod clar CNNProm în toate seturile de date cu toate valorile de evaluare. Mai precis, DeePromoter îmbunătățește precizia, rechemarea și MCC în cazul setului de date TATA uman cu 0,18, 0,04 și, respectiv, 0,26. În cazul setului de date uman non-TATA, DeePromoter îmbunătățește precizia cu 0,39, rechemarea cu 0,12 și MCC cu 0,66. În mod similar, DeePromoter îmbunătățește precizia și MCC în cazul setului de date TATA al mouse-ului cu 0,24 și respectiv 0,31. În cazul setului de date non-TATA al mouse-ului, DeePromoter îmbunătățește precizia cu 0,37, rechemarea cu 0,04 și MCC cu 0,65. Aceste rezultate confirmă faptul că CNNProm nu reușește să respingă secvențele negative cu promotorul TATA, prin urmare, are un fals pozitiv ridicat. Pe de altă parte, modelele noastre sunt capabile să facă față acestor cazuri cu mai mult succes, iar rata fals pozitivă este mai mică în comparație cu CNNProm.

Tabelul 2. Comparația DeePromoter cu starea de -metoda artei.
Pentru analize suplimentare, studiem efectul alternării nucleotidelor la fiecare poziție asupra scorului de ieșire. Ne concentrăm asupra regiunii –40 și 10 pb, deoarece găzduiește cea mai importantă parte a secvenței promotorului. Pentru fiecare secvență promotor din setul de testare, efectuăm scanarea mutațională de calcul pentru a evalua efectul mutării fiecărei baze a subsecvenței de intrare (150 substituții pe intervalul –40 ~ 10 bp subsecvență). Acest lucru este ilustrat în Figurile 6, 7 pentru seturile de date TATA pentru om și respectiv pentru șoarece. Culoarea albastră reprezintă o scădere a scorului de ieșire din cauza mutației, în timp ce culoarea roșie reprezintă creșterea scorului din cauza mutației. Observăm că modificarea nucleotidelor la C sau G în regiunea –30 și –25 bp reduce semnificativ scorul de ieșire. Această regiune este TATA-box, care este un motiv funcțional foarte important în secvența promotorului. Astfel, modelul nostru este capabil să găsească cu succes importanța acestei regiuni. În restul pozițiilor, nucleotidele C și G sunt mai preferabile decât A și T, mai ales în cazul șoarecelui. Acest lucru poate fi explicat prin faptul că regiunea promotoră are mai multe nucleotide C și G decât A și T (Shi și Zhou, 2006).

Figura 6. Harta saliency a regiunii –40 bp la 10 bp, care include caseta TATA, în cazul secvențelor promotor TATA uman.

Figura 7. Harta de sănătate a regiunii –40 bp la 10 bp, care include caseta TATA, în cazul secvențelor promotor TATA ale mouse-ului.
Concluzie
Predicția exactă a secvențelor promotor este esențială pentru înțelegerea mecanismului de bază al procesului de reglare a genelor. În această lucrare, am dezvoltat DeePromoter – care se bazează pe o combinație de rețea neuronală de convoluție și LSTM bidirecțional – pentru a prezice secvențele scurte ale promotorului eucariot în cazul omului și al șoarecelui atât pentru promotorul TATA, cât și pentru cel care nu este TATA. Componenta esențială a acestei lucrări a fost de a depăși problema preciziei scăzute (rata fals pozitivă ridicată) observată în instrumentele dezvoltate anterior datorită dependenței de unele caracteristici / motive evidente din secvență atunci când se clasifică secvențele promotor și non-promotor. În această lucrare, am fost deosebit de interesați să construim un set negativ dur care să conducă modelele spre explorarea secvenței pentru trăsături profunde și relevante în loc să distingem doar secvențele promotor și non-promotor pe baza existenței unor motive funcționale. Principalele avantaje ale utilizării DeePromoter este că reduce semnificativ numărul de predicții fals pozitive, obținând în același timp o precizie ridicată în seturile de date provocatoare. DeePromoter a depășit metoda anterioară nu numai în ceea ce privește performanța, ci și în depășirea problemei predicțiilor false pozitive ridicate. Se preconizează că acest cadru ar putea fi util în aplicațiile legate de droguri și în mediul academic.
Contribuțiile autorului
MO și ZL au pregătit setul de date, au conceput algoritmul și au realizat experimentul și analiză. MO și HT au pregătit serverul web și au scris manuscrisul cu sprijinul ZL și KC. Toți autorii au discutat rezultatele și au contribuit la manuscrisul final.
Finanțare
Această cercetare a fost susținută de Programul de cercetare a creierului al Fundației Naționale de Cercetare (NRF) finanțat de guvernul coreean ( MSIT) (Nr. NRF-2017M3C7A1044815).
Declarație privind conflictul de interese
Autorii declară că cercetarea a fost efectuată în absența oricărei relații comerciale sau financiare care ar putea fi interpretată ca un potențial conflict de interese.
Bharanikumar, R., Premkumar, KAR și Palaniappan, A. (2018). Promoterpredict: modelarea pe secvență a puterii promotorului escherichia coli σ70 produce dependență logaritmică între puterea și secvența promotorului. PeerJ 6: e5862. doi: 10.7717 / peerj.5862
PubMed Abstract | Text integral CrossRef | Google Scholar
Glorot, X., Bordes, A. și Bengio, Y. (2011). „Rețele neuronale de redresare redusă”, în Proceedings of the XIV Conference International on Artificial Intelligence and Statistics, (Fort Lauderdale, FL:) 315–323.
Google Scholar
Hutchinson, G. (1996). Predicția regiunilor promotor al vertebratelor utilizând analiza diferențială a frecvenței hexamerului. Bioinformatică 12, 391-398.
PubMed Abstract | Google Scholar
Kingma, DP și Ba, J. (2014). Adam: o metodă de optimizare stocastică. arXiv preprint arXiv: 1412.6980.
Google Scholar
Knudsen, S. (1999). Promotor2. 0: pentru recunoașterea secvențelor promotorului polii. Bioinformatics 15, 356-361.
PubMed Abstract | Google Scholar
Ponger, L. și Mouchiroud, D. (2002). Cpgprod: identificarea insulelor cpg asociate cu site-urile de pornire a transcrierii în secvențe mari de mamifere genomice. Bioinformatică 18, 631–633. doi: 10.1093 / bioinformatics / 18.4.631
PubMed Abstract | Text integral CrossRef | Google Scholar
Quang, D. și Xie, X. (2016). Danq: o rețea neuronală profundă recurentă și recurentă hibridă pentru cuantificarea funcției secvențelor ADN. Acizi nucleici Res. 44, e107 – e107. doi: 10.1093 / nar / gkw226
PubMed Abstract | Text integral CrossRef | Google Scholar
Umarov, R. K. și Solovyev, V. V. (2017). Recunoașterea promotorilor procariote și eucariote utilizând rețele neuronale de învățare profundă convoluțională. PLoS ONE 12: e0171410. doi: 10.1371 / journal.pone.0171410
PubMed Abstract | Text integral CrossRef | Google Scholar