Gränser i genetik
Inledning
Arrangörer är nyckelelementen som tillhör icke-kodande regioner i genomet. De styr till stor del aktivering eller förtryck av generna. De är belägna nära och uppströms genens transkriptionsstartplats (TSS). En genpromotorflankerande region kan innehålla många viktiga korta DNA-element och motiv (5 och 15 baser långa) som fungerar som igenkänningsställen för proteinerna som ger korrekt initiering och reglering av transkription av nedströmsgenen (Juven-Gershon et al., 2008). Initieringen av gentranskript är det mest grundläggande steget i regleringen av genuttryck. Promotorkärnan är en minimal sträcka av DNA-sekvens som bildar TSS och är tillräcklig för att direkt initiera transkriptionen. Längden på kärnpromotorn varierar typiskt mellan 60 och 120 baspar (bp).
TATA-rutan är en promotorsekvens som indikerar för andra molekyler var transkription börjar. Den fick namnet ”TATA-box” eftersom dess sekvens kännetecknas av att upprepa T- och A-baspar (TATAAA) (Baker et al., 2003). De allra flesta studier på TATA-boxen har utförts på människa, jäst, och Drosophila-genom har emellertid liknande element hittats i andra arter såsom archaea och forntida eukaryoter (Smale och Kadonaga, 2003). I mänskliga fall har 24% av gener promotorregioner som innehåller TATA-box (Yang et al., 2007 I eukaryoter ligger TATA-box vid ~ 25 bp uppströms om TSS (Xu et al., 2016). Den kan definiera transkriptionsriktningen och indikerar också DNA-strängen som ska läsas. Proteiner som kallas transkriptionsfaktorer binder till flera icke-kodande regioner inklusive TATA-box och rekryterar ett enzym som kallas RNA-polymeras, som syntetiserar RNA från DNA.
På grund av promotorernas viktiga roll i gentranskription blir exakt förutsägelse av promotorsidor ett nödvändigt steg i genuttryck, tolkning av mönster och byggande och förståelse funktionaliteten hos genetiska regleringsnätverk. Det fanns olika biologiska experiment för identifiering av promotorer såsom mutationsanalys (Matsumine et al., 1998) och immunutfällningsanalyser (Kim et al., 2004; Dahl och Collas, 2008). Dessa metoder var dock både dyra och tidskrävande. Nyligen, med utvecklingen av nästa generations sekvensering (NGS) (Behjati och Tarpey, 2013) har fler gener från olika organismer sekvenserats och deras genelement har undersökts beräkningsmässigt (Zhang et al., 2011). Å andra sidan har innovationen av NGS-teknik resulterat i ett dramatiskt fall av kostnaden för hela genom-sekvenseringen, vilket innebär att mer sekvenseringsdata finns tillgängliga. Datatillgängligheten lockar forskare att utveckla beräkningsmodeller för promotor förutsägelse uppgift. Det är dock fortfarande en ofullständig uppgift och det finns ingen effektiv programvara som kan förutsäga promotorer exakt.
Promotorprediktorer kan kategoriseras baserat på den använda metoden i tre grupper, nämligen signalbaserad strategi, innehållsbaserad strategi och den GpG-baserade metoden. Signalbaserade prediktorer fokuserar på promotorelement relaterade till RNA-polymerasbindningsställe och ignorerar icke-elementdelarna i sekvensen. Som ett resultat var förutsägelsens noggrannhet svag och inte tillfredsställande. Exempel på signalbaserade prediktorer inkluderar: PromoterScan (Prestridge, 1995) som använde de extraherade funktionerna i TATA-rutan och en viktad matris av transkriptionsfaktorbindningsställen med en linjär diskriminator för att klassificera promotorsekvenser från icke-promotor-sådana; Promoter2.0 (Knudsen, 1999) som extraherade funktionerna från olika lådor som TATA-Box, CAAT-Box och GC-Box och skickade dem till artificiella neurala nätverk (ANN) för klassificering; NNPP2.1 (Reese, 2001) som använde initiatorelementet (Inr) och TATA-Box för extraktion av funktioner och ett tidsfördröjningsneuralt nätverk för klassificering och Down and Hubbard (2002) som använde TATA-Box och använde en relevansvektormaskiner (RVM) som klassificerare. Innehållsbaserade prediktorer förlitade sig på att räkna frekvensen av k-mer genom att köra ett fönster med k-längd över sekvensen. Dessa metoder ignorerar emellertid den rumsliga informationen för basparen i sekvenserna. Exempel på innehållsbaserade prediktorer inkluderar: PromFind (Hutchinson, 1996) som använde k-mer-frekvensen för att utföra förutsägelsen av hexamerpromotorn; PromoterInspector (Scherf et al., 2000) som identifierade regionerna innehållande promotorer baserat på ett gemensamt genomiskt sammanhang av polymeras II-promotorer genom att söka efter specifika egenskaper definierade som motiv med variabel längd; MCPromoter1.1 (Ohler et al., 1999) som använde en enda interpolerad Markov-kedja (IMC) av 5: e ordningen för att förutsäga promotorsekvenser.Slutligen använde GpG-baserade prediktorer placeringen av GpG-öarna som promotorregion eller den första exonregionen i de mänskliga generna innehåller vanligtvis GpG-öar (Ioshikhes och Zhang, 2000; Davuluri et al., 2001; Lander et al., 2001; Ponger och Mouchiroud, 2002). Emellertid innehåller endast 60% av promotorerna GpG-öar, därför har förutsägelsesnoggrannheten för denna typ av prediktorer aldrig överskridit 60%.
Nyligen har sekvensbaserade tillvägagångssätt använts för promotorprognos. Yang et al. (2017) använde olika funktionsextraktionsstrategier för att fånga den mest relevanta sekvensinformationen för att förutsäga interaktioner mellan förstärkare och promotorer. Lin et al. (2017) föreslog en sekvensbaserad prediktor, med namnet ”iPro70-PseZNC”, för identifiering av sigma70-promotor i prokaryoten. På samma sätt har Bharanikumar et al. (2018) föreslog PromoterPredict för att förutsäga styrkan hos Escherichia coli-promotorer baserat på ett dynamiskt multipelt regressionssätt där sekvenserna representerades som positionsviktmatriser (PWM). Kanhere och Bansal (2005) använde skillnaderna i DNA-sekvensstabilitet mellan promotorsekvenser och icke-promotorsekvenser för att särskilja dem. Xiao et al. (2018) introducerade en tvåskiktsprediktor som heter iPSW (2L) -PseKNC för promotorsekvensidentifiering såväl som promotorernas styrka genom att extrahera hybridfunktioner från sekvenserna.
Alla ovannämnda prediktorer kräver domän- kunskap för att handgjorda funktionerna. Å andra sidan möjliggör djupinlärningsbaserade metoder att bygga effektivare modeller med rådata (DNA / RNA-sekvenser) direkt. Djupt konvolutionellt neuralt nätverk uppnådde toppmoderna resultat i utmanande uppgifter som att bearbeta bild, video, ljud och tal (Krizhevsky et al., 2012; LeCun et al., 2015; Schmidhuber, 2015; Szegedy et al. , 2015). Dessutom applicerades den framgångsrikt i biologiska problem som DeepBind (Alipanahi et al., 2015), DeepCpG (Angermueller et al., 2017), val av förgreningspunkt (Nazari et al., 2018), alternativ splicing sites förutsägelse (Oubounyt et al., 2018), 2 ”-Ometyleringsställen förutsägelse (Tahir et al., 2018), DNA-sekvenskvantifiering (Quang och Xie, 2016), humant protein subcellulär lokalisering (Wei et al., 2018), etc. CNN fick nyligen stor uppmärksamhet i uppgiften för promotorigenkänning. Mycket nyligen introducerade Umarov och Solovyev (2017) CNNprom för korta promotorsekvenser, denna CNN-baserade arkitektur uppnådde höga resultat när det gäller att klassificera promotors och icke-promotorsekvenser. Därefter förbättrades denna modell av Qian et al. (2018) där författarna använde support vector machine (SVM) klassificerare för att inspektera de viktigaste promotorsekvenselementen. Därefter hölls de mest inflytelserika elementen okomprimerade medan de mindre viktiga komprimerades. Denna process resulterade i bättre prestanda. Nyligen föreslogs lång modell för identifiering av promotorer av Umarov et al. (2019) där författarna fokuserade på identifieringen av TSS-position.
I alla ovannämnda verk extraherades den negativa uppsättningen från icke-promotorregioner i genomet. Att veta att promotorsekvenserna är rika uteslutande av specifika funktionella element såsom TATA-box som ligger vid –30 ~ –25 bp, GC-Box som ligger vid –110 ~ –80 bp, CAAT-Box som ligger vid – 80 ~ –70 bp, etc. Detta resulterar i hög klassificeringsnoggrannhet på grund av enorm skillnad mellan de positiva och negativa proverna när det gäller sekvensstruktur. Dessutom blir klassificeringsuppgiften enkel att uppnå, till exempel kommer CNN-modellerna bara att förlita sig på närvaron eller frånvaron av vissa motiv i deras specifika positioner för att fatta beslut om sekvenstyp. Således har dessa modeller mycket låg precision / känslighet (högt falskt positivt) när de testas på genomiska sekvenser som har promotormotiv men de är inte promotorsekvenser. Det är välkänt att det finns fler TATAAA-motiv i genomet än de som tillhör promotorregionerna. Exempelvis innehåller DNA-sekvensen för den humana kromosomen 1, ftp://ftp.ensembl.org/pub/release-57/fasta/homo_sapiens/dna/, 151 656 TATAAA-motiv. Det är mer än det ungefärliga maximala antalet gener i det totala humana genomet. Som en illustration av denna fråga märker vi att när vi testar dessa modeller på icke-promotorsekvenser som har TATA-box, klassificerar de felaktigt de flesta av dessa sekvenser. Därför, för att generera en robust klassificering, bör den negativa uppsättningen väljas noggrant eftersom den bestämmer de funktioner som klassificeraren kommer att använda för att diskriminera klasserna. Vikten av denna idé har demonstrerats i tidigare verk som (Wei et al., 2014). I detta arbete behandlar vi huvudsakligen denna fråga och föreslår ett tillvägagångssätt som integrerar några av de positiva klassfunktionella motiv i den negativa klassen för att minska modellens beroende av dessa motiv.Vi använder en CNN kombinerad med LSTM-modell för att analysera sekvensegenskaper hos humana och mus-TATA och icke-TATA eukaryota promotorer och bygga beräkningsmodeller som exakt kan diskriminera korta promotorsekvenser från icke-promotor-sådana.
2.1. Dataset
Datauppsättningarna, som används för att träna och testa den föreslagna promotorprediktorn, samlas in från människa och mus. De innehåller två distinkta klasser av promotorerna, nämligen TATA-promotorer (dvs. sekvenserna som innehåller TATA-box) och icke-TATA-promotorer. Dessa datamängder byggdes från Eukaryotic Promoter Database (EPDnew) (Dreos et al., 2012). EPDnew är ett nytt avsnitt under den välkända EPD-datasetet (Périer et al., 2000) som antecknas en icke-redundant samling av eukaryota POL II-promotorer där transkriptionsstartplats har bestämts experimentellt. Det ger promotorer av hög kvalitet jämfört med ENSEMBL-promotorsamling (Dreos et al., 2012) och det är offentligt tillgängligt på https://epd.epfl.ch//index.php. Vi laddade ner TATA- och icke-TATA-promotors genomiska sekvenser för varje organism från EPDnew. Denna operation resulterade i att man erhöll fyra promotordatamängder, nämligen: Human-TATA, Human-non-TATA, Mouse-TATA och Mouse-non-TATA. För var och en av dessa datamängder konstrueras en negativ uppsättning (icke-promotorsekvenser) med samma storlek som den positiva baserat på det föreslagna tillvägagångssättet som beskrivs i följande avsnitt. Detaljerna om antalet promotorsekvenser för varje organism ges i tabell 1. Alla sekvenser har en längd av 300 bp och extraherades från -249 ~ + 50 bp (+1 avser TSS-position). Som kvalitetskontroll använde vi femfaldig korsvalidering för att bedöma den föreslagna modellen. I det här fallet används tre gånger för träning, 1 gånger används för validering och återstående vik används för testning. Således tränas den föreslagna modellen fem gånger och den totala prestandan för 5-faldet beräknas.

Tabell 1. Statistik över de fyra datamängder som används i denna studie.
2.2. Negativ datakonstruktion
För att träna en modell som exakt kan utföra promotor- och icke-promotorsekvensklassificering, måste vi välja den negativa uppsättningen (icke-promotorsekvenser) noggrant. Denna punkt är avgörande för att göra en modell som kan generalisera väl och därför kunna behålla sin precision när den utvärderas på mer utmanande datamängder. Tidigare arbeten, såsom (Qian et al., 2018), konstruerade negativ uppsättning genom att slumpmässigt välja fragment från genom-icke-promotorregioner. Uppenbarligen är detta tillvägagångssätt inte helt rimligt, för om det inte finns någon skärningspunkt mellan positiva och negativa uppsättningar. Således kommer modellen enkelt att hitta grundläggande funktioner för att separera de två klasserna. Till exempel kan TATA-motiv hittas i alla positiva sekvenser vid en specifik position (normalt 28 bp uppströms om TSS, mellan –30 och –25 pb i vår dataset). Därför skapar du slumpmässigt negativ uppsättning som inte innehåller detta motiv höga prestanda i denna dataset. Modellen misslyckas dock med att klassificera negativa sekvenser som har TATA-motiv som promotorer. Kort sagt är den största bristen i detta tillvägagångssätt att när man tränar en djup inlärningsmodell lär den sig bara att diskriminera de positiva och negativa klasserna baserat på närvaron eller frånvaron av några enkla funktioner på specifika positioner, vilket gör dessa modeller omöjliga. I det här arbetet strävar vi efter att lösa denna fråga genom att skapa en alternativ metod för att härleda den negativa uppsättningen från den positiva.
Vår metod bygger på det faktum att närhelst funktionerna är vanliga mellan negativa och positiv klass, tenderar modellen att när man fattar beslutet ignorera eller minska dess beroende av dessa funktioner (dvs. tilldela dessa funktioner låga vikter). Istället tvingas modellen att söka efter djupare och mindre uppenbara funktioner. Deep learning-modeller lider i allmänhet av långsam konvergens när de tränar på denna typ av data. Denna metod förbättrar dock robustheten i modellen och säkerställer generalisering. Vi rekonstruerar den negativa uppsättningen enligt följande. Varje positiv sekvens genererar en negativ sekvens. Den positiva sekvensen är uppdelad i 20 sekvenser. Därefter väljs 12 sekvenser slumpmässigt och ersätts slumpmässigt. De återstående åtta sekvenserna sparas. Denna process illustreras i figur 1. Att tillämpa denna process på den positiva uppsättningen resulterar i nya icke-promotorsekvenser med konserverade delar från promotorsekvenser (de oförändrade sekvenserna, 8 sekvenser av 20). Dessa parametrar möjliggör generering av en negativ uppsättning som har 32 och 40% av dess sekvenser som innehåller konserverade delar av promotorsekvenser. Detta förhållande har visat sig vara optimalt för att ha en robust promotorprediktor som förklaras i avsnitt 3.2.Eftersom de konserverade delarna upptar samma positioner i de negativa sekvenserna är de uppenbara motiv som TATA-box och TSS nu vanliga mellan de två uppsättningarna med ett förhållande på 32 ~ 40%. Sekvenslogotyperna för de positiva och negativa uppsättningarna för både humana och mus-TATA-promotordata visas i figurerna 2, 3 respektive. Det kan ses att de positiva och de negativa uppsättningarna delar samma grundmotiv vid samma positioner som TATA-motivet vid positionen -30 och –25 bp och TSS vid positionen +1 bp. Därför är utbildningen mer utmanande men den resulterade modellen generaliserar väl.
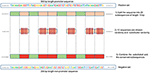
Figur 1. Illustration av den negativa konstruktionsmetoden. Grön representerar slumpmässigt konserverade sekvenser medan rött representerar slumpmässigt valda och substituerade.

Figur 2. Sekvenslogotypen i human TATA-promotor för både positiv uppsättning (A) och negativ uppsättning (B). Diagrammen visar bevarandet av funktionella motiv mellan de två uppsättningarna.

Figur 3. Sekvenslogotypen i mus TATA-promotor för både positiv uppsättning (A) och negativ uppsättning (B). Diagrammen visar bevarandet av funktionella motiv mellan de två uppsättningarna.
2.3. De föreslagna modellerna
Vi föreslår en djup inlärningsmodell som kombinerar fällningsskikt med återkommande lager som visas i figur 4. Den accepterar en enda rå genomisk sekvens, S = {N1, N2,…, Nl} där N ∈ {A, C, G, T} och l är längden på ingångssekvensen, som inmatning och utmatning av en verkligt värderad poäng. Ingången är enkodad och representerad som en endimensionell vektor med fyra kanaler. Längden på vektorn l = 300 och de fyra kanalerna är A, C, G och T och representeras som (1 0 0 0), (0 1 0 0), (0 0 1 0), (0 0 0 1 ), respektive. För att välja den bäst presterande modellen har vi använt rutnätssökningsmetod för att välja de bästa hyperparametrarna. Vi har provat olika arkitekturer som CNN ensam, LSTM ensam, BiLSTM ensam, CNN kombinerat med LSTM. De inställda hyperparametrarna är antalet fällningsskikt, kärnstorlek, antal filter i varje skikt, storleken på maxlagringslagret, bortfallssannolikhet och enheterna i Bi-LSTM-skiktet.
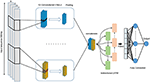
Figur 4. Arkitekturen för den föreslagna DeePromoter-modellen.
Den föreslagna modellen börjar med flera fällningsskikt som är inriktade parallellt och hjälper till att lära sig de viktiga motiven för inmatningssekvenserna med olika fönsterstorlek. Vi använder tre fällningsskikt för icke-TATA-promotor med fönsterstorlekar 27, 14 och 7 och två fällningsskikt för TATA-promotorer med fönsterstorlekar 27, 14. Alla fällningsskikt följs av ReLU-aktiveringsfunktion (Glorot et al. , 2011), ett maximalt poollager med en fönsterstorlek på 6 och ett dropout-lager med en sannolikhet 0,5. Därefter sammanfogas utgångarna från dessa lager och matas in i ett dubbelriktat långt korttidsminne (BiLSTM) (Schuster och Paliwal, 1997) med 32 noder för att fånga beroenden mellan de inlärda motiven från konvolutionslagren. De inlärda funktionerna efter BiLSTM planas ut och följs av bortfall med en sannolikhet på 0,5. Sedan lägger vi till två helt anslutna lager för klassificering. Den första har 128 noder och följt av ReLU och bortfall med en sannolikhet på 0,5 medan det andra skiktet används för förutsägelse med en nod- och sigmoid-aktiveringsfunktion. BiLSTM låter informationen bestå och lära sig långsiktiga beroenden av sekventiella prover som DNA och RNA. Detta uppnås genom LSTM-strukturen som består av en minnescell och tre grindar som kallas ingångs-, utgångs- och glömgrindar. Dessa grindar är ansvariga för att reglera informationen i minnescellen. Dessutom ökar användningen av LSTM-modulen nätverksdjupet medan antalet nödvändiga parametrar förblir lågt. Att ha ett djupare nätverk möjliggör extrahering av mer komplexa funktioner och detta är huvudmålet för våra modeller eftersom den negativa uppsättningen innehåller hårda prover.
Keras-ramverket används för att konstruera och träna de föreslagna modellerna (Chollet F. et. al., 2015). Adam optimizer (Kingma och Ba, 2014) används för att uppdatera parametrarna med en inlärningshastighet på 0,001. Satsstorleken är inställd på 32 och antalet epoker är inställt på 50. Tidigt stopp stoppas baserat på valideringsförlust.
Resultat och diskussion
3.1. Prestationsåtgärder
I detta arbete använder vi de allmänt använda utvärderingsmätvärdena för att utvärdera prestanda för de föreslagna modellerna.Dessa mätvärden är precision, återkallande och Matthew korrelationskoefficient (MCC), och de definieras enligt följande:
Där TP är sant positivt och representerar korrekt identifierade promotorsekvenser, är TN sant negativ och representerar korrekt avvisade promotorsekvenser, FP är falskt positivt och representerar felaktigt identifierade promotorsekvenser och FN är falskt negativ och representerar felaktigt avvisade promotorsekvenser.
3.2. Effekt av den negativa uppsättningen
När vi analyserade tidigare publicerade verk för promotorsekvensidentifiering märkte vi att prestationen för dessa verk i hög grad beror på sättet att förbereda den negativa datasetet. De presterade mycket bra på de datauppsättningar som de har förberett, men de har ett högt falskt positivt förhållande när de utvärderas i en mer utmanande dataset som inkluderar icke-prompter-sekvenser som har gemensamma motiv med promotorsekvenser. Till exempel, i fallet med TATA-promotordataset, kommer de slumpmässigt genererade sekvenserna inte att ha TATA-motiv vid positionen -30 och –25 bp vilket i sin tur gör uppgiften att klassificera enklare. Med andra ord berodde deras klassificerare på närvaron av TATA-motiv för att identifiera promotorsekvensen och som ett resultat var det lätt att uppnå hög prestanda på de datamängder de har förberett. Men deras modeller misslyckades dramatiskt när de behandlade negativa sekvenser som innehöll TATA-motiv (hårda exempel). Precisionen sjönk när den falskt positiva frekvensen ökade. Enkelt klassificerade de dessa sekvenser som positiva promotorsekvenser. En liknande analys gäller för andra promotormotiv. Därför är det huvudsakliga syftet med vårt arbete inte bara att uppnå hög prestanda på en specifik dataset utan också att förbättra modellförmågan att generalisera väl genom att träna på en utmanande dataset.
För att mer illustrera denna punkt, tränar vi och testa vår modell på TATA-promotorns datamängder för människor och mus med olika metoder för förberedelse av negativa uppsättningar. Det första experimentet utförs med användning av slumpmässigt samplade negativa sekvenser från icke-kodande regioner i genomet (d.v.s. liknar det tillvägagångssätt som användes i tidigare arbeten). Anmärkningsvärt är att vår föreslagna modell uppnår nästan perfekt förutsägelsesnoggrannhet (precision = 99%, återkallelse = 99%, Mcc = 98%) och (precision = 99%, återkallelse = 98%, Mcc = 97%) för både människa respektive mus . Dessa höga resultat förväntas, men frågan är om den här modellen kan upprätthålla samma prestanda när den utvärderas i en dataset som har hårda exempel. Svaret, baserat på analys av tidigare modeller, är nej. Det andra experimentet utförs med vår föreslagna metod för att förbereda datamängden som förklaras i avsnitt 2.2. Vi förbereder de negativa uppsättningarna som innehåller konserverade TATA-boxar med olika procentsatser som 12, 20, 32 och 40% och målet är att minska klyftan mellan precisionen och återkallelsen. Detta säkerställer att vår modell lär sig mer komplexa funktioner snarare än att bara lära sig närvaron eller frånvaron av TATA-box. Som visas i figurerna 5A stabiliseras B modellen i förhållandet 32 ~ 40% för både TATA-promotormängder för människor och mus.
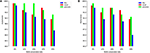
Figur 5. Effekten av olika konserveringsförhållanden för TATA-motiv i den negativa uppsättningen på prestanda vid TATA-promotordataset för både mänsklig (A) och mus (B) .
3.3. Resultat och jämförelse
Under de senaste åren har massor av förutsägelsesverktyg för regioner föreslagits (Hutchinson, 1996; Scherf et al., 2000; Reese, 2001; Umarov och Solovyev, 2017). Några av dessa verktyg är dock inte offentligt tillgängliga för testning och vissa av dem kräver mer information förutom de råa genomiska sekvenserna. I den här studien jämför vi prestanda för våra föreslagna modeller med nuvarande toppmoderna verk, CNNProm, som föreslogs av Umarov och Solovyev (2017) som visas i tabell 2. Generellt sett är de föreslagna modellerna, DeePromoter, klart överträffa CNNProm i alla datamängder med alla utvärderingsmått. Mer specifikt förbättrar DeePromoter precision, återkallande och MCC i fallet med mänsklig TATA-dataset med 0,18, 0,04 respektive 0,26. När det gäller humant icke-TATA-dataset, förbättrar DeePromoter precisionen med 0,39, återkallandet med 0,12 och MCC med 0,66. På samma sätt förbättrar DeePromoter precisionen och MCC i fallet med mus-TATA-dataset med 0,24 respektive 0,31. När det gäller mus som inte är TATA-dataset förbättrar DeePromoter precisionen med 0,37, återkallandet med 0,04 och MCC med 0,65. Dessa resultat bekräftar att CNNProm misslyckas med att avvisa negativa sekvenser med TATA-promotor, därför har den hög falskt positiv. Å andra sidan kan våra modeller hantera dessa fall mer framgångsrikt och falskt positivt är lägre jämfört med CNNProm.

Tabell 2. Jämförelse av DeePromoter med tillståndet för -teknik-metoden.
För ytterligare analyser studerar vi effekten av alternerande nukleotider vid varje position på utdatapoängen. Vi fokuserar på regionen –40 och 10 bp eftersom den är värd för den viktigaste delen av promotorsekvensen. För varje promotorsekvens i testuppsättningen utför vi beräkningsmutationsskanning för att utvärdera effekten av att mutera varje bas av ingångssekvensen (150 substitutioner på intervallet –40 ~ 10 bp efterföljande). Detta illustreras i figurerna 6, 7 för TATA-datamängder för människor respektive mus. Blå färg representerar en nedgång i utdatapoängen på grund av mutation medan den röda färgen representerar ökningen av poängen på grund av mutation. Vi märker att om du ändrar nukleotiderna till C eller G i regionen –30 och –25 bp minskar utdatapoängen avsevärt. Denna region är TATA-box som är ett mycket viktigt funktionellt motiv i promotorsekvensen. Således kan vår modell framgångsrikt hitta vikten av denna region. I övriga positioner är C- och G-nukleotider mer föredragna än A och T, särskilt i fallet med musen. Detta kan förklaras av det faktum att promotorregionen har mer C- och G-nukleotider än A och T (Shi och Zhou, 2006).

Figur 6. Markeringskartan för regionen –40 bp till 10 bp, som inkluderar TATA-rutan, i fallet med humana TATA-promotorsekvenser.

Figur 7. Markeringskartan för regionen –40 bp till 10 bp, som inkluderar TATA-rutan, i fallet med mus TATA-promotorsekvenser.
Slutsats
Noggrann förutsägelse av promotorsekvenser är väsentlig för att förstå den underliggande mekanismen för genregleringsprocessen. I det här arbetet utvecklade vi DeePromoter -som är baserad på en kombination av neurologiskt nätverk av konvolution och dubbelriktad LSTM- för att förutsäga de korta eukaryot-promotorsekvenserna i fall av människa och mus för både TATA och icke-TATA-promotor. Den väsentliga komponenten i detta arbete var att övervinna frågan om låg precision (hög falsk positiv frekvens) som noterats i de tidigare utvecklade verktygen på grund av beroende av några uppenbara funktioner / motiv i sekvensen vid klassificering av promotorsekvenser och icke-promotorsekvenser. I det här arbetet var vi särskilt intresserade av att konstruera en hård negativ uppsättning som driver modellerna mot att utforska sekvensen för djupa och relevanta funktioner istället för att bara skilja på promotorsekvenser och icke-promotorsekvenser baserat på förekomsten av vissa funktionella motiv. De största fördelarna med att använda DeePromoter är att det avsevärt minskar antalet falska positiva förutsägelser samtidigt som det uppnås hög noggrannhet i utmanande datamängder. DeePromoter överträffade den tidigare metoden inte bara i prestanda utan också i att övervinna frågan om höga falska positiva förutsägelser. Det beräknas att detta ramverk kan vara till hjälp i läkemedelsrelaterade applikationer och den akademiska världen.
Författarbidrag
MO och ZL förberedde datauppsättningen, utformade algoritmen och genomförde experimentet och analys. MO och HT förberedde webservern och skrev manuskriptet med stöd från ZL och KC. Alla författare diskuterade resultaten och bidrog till det slutliga manuskriptet.
Finansiering
Denna forskning stöddes av hjärnforskningsprogrammet från National Research Foundation (NRF) finansierat av den koreanska regeringen ( MSIT) (nr NRF-2017M3C7A1044815).
Uttalande av intressekonflikter
Författarna förklarar att forskningen utfördes i avsaknad av kommersiella eller ekonomiska relationer som kan tolkas en potentiell intressekonflikt.
Bharanikumar, R., Premkumar, KAR och Palaniappan, A. (2018). Promoterpredict: sekvensbaserad modellering av escherichia coli σ70-promotorstyrka ger logaritmiskt beroende mellan promotorstyrka och sekvens. PeerJ 6: e5862. doi: 10.7717 / peerj.5862
PubMed Abstract | CrossRef Fulltext | Google Scholar
Glorot, X., Bordes, A. och Bengio, Y. (2011). ”Deep sparse rectifier neural networks,” i Proceedings of the Fourteenth International Conference on Artificial Intelligence and Statistics, (Fort Lauderdale, FL:) 315–323.
Google Scholar
Hutchinson, G. (1996). Förutsägelse av regioner med ryggradsdrivande promotorer med differentiell hexamerfrekvensanalys. Bioinformatik 12, 391–398.
PubMed Abstract | Google Scholar
Kingma, DP och Ba, J. (2014). Adam: en metod för stokastisk optimering. arXiv preprint arXiv: 1412.6980.
Google Scholar
Knudsen, S. (1999). Promoter2. 0: för igenkänning av polii-promotorsekvenser. Bioinformatik 15, 356–361.
PubMed Abstract | Google Scholar
Ponger, L. och Mouchiroud, D. (2002). Cpgprod: identifiering av cpg-öar associerade med transkriptionsstartplatser i stora genomiska däggdjurssekvenser. Bioinformatik 18, 631–633. doi: 10.1093 / bioinformatics / 18.4.631
PubMed Abstract | CrossRef Fulltext | Google Scholar
Quang, D. och Xie, X. (2016). Danq: ett hybridkonvolutions- och återkommande djupt neurala nätverk för att kvantifiera funktionen hos dna-sekvenser. Nucleic Acids Res. 44, e107 – e107. doi: 10.1093 / nar / gkw226
PubMed Abstract | CrossRef Fulltext | Google Scholar
Umarov, R. K. och Solovyev, V. V. (2017). Erkännande av prokaryota och eukaryota initiativtagare med hjälp av omvälvande djupinlärande neurala nätverk. PLoS ONE 12: e0171410. doi: 10.1371 / journal.pone.0171410
PubMed Abstract | CrossRef Fulltext | Google Scholar